The influence of NH4+ ions on the corrosion behavior of AZ31 magnesium alloy was investigated by immersion test, hydrogen evolution, electrochemical methods and morphology observation. The results demonstrate the acceleration effect of NH4+ on corrosion of AZ31 magnesium alloy due to the disruption of protective MgO film in NH4+-containing solution. The loose and cracked corrosion products of AZ31 magnesium alloy in NH4+-containing solutions are mainly composed of (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O and Mg5(CO3)4(OH)2·5H2O. When the NH4+ concentration is lower than 0.01 M, knife-cut like corrosion occurs in some active area of the surface due to the partial dissolution of MgO layer. As the NH4+ concentration is increased to 0.1 M, the MgO layer is completely disrupted, resulting in the occurrence of uniform corrosion. Both cathodic and anodic reactions are accelerated by NH4+ ions, while the effect of original pH values on the reaction kinetics can be neglected in NH4+-containing solutions.
Hui Pan, Liwei Wang, Yi Lin, Feng Ge, Kang Zhao, Xin Wang, Zhongyu Cui. Mechanistic study of ammonium-induced corrosion of AZ31 magnesium alloy in sulfate solution. Journal of Materials Science & Technology[J], 2020, 54(0): 1-13 DOI:10.1016/j.jmst.2020.02.074
1. Introduction
For decades, the rapid development of industry has aggravated air pollution in several developing countries [[1], [2], [3]]. The air pollution not only does harm to human health, but also affects the corrosion of some metallic materials because of the presence of particulate matter (PM). It is well-known that water-soluble inorganic ions (WSIIs) are important ingredients in PM2.5 mass. Zhang et al. [4] reported that the main WSIIs are sulfate (SO42-), ammonium (NH4+), chloride (Cl-), and nitrate ions (NO3-). When pollution particles containing these ions come into contact with the metal surface, they can easily absorb water from the surroundings and form a thin electrolyte layer covering the metal surface [5]. This creates an aggressive environment that attacks metallic materials. Therefore, it is meaningful to study the influence of these ions on the corrosion of metallic materials.
Magnesium (Mg) alloys including AZ and AM series are widely used in automotive, aircraft, electronic equipment, and other lightweight fields [[6], [7], [8], [9]]. However, the poor corrosion resistance is the most important property that limits the application of these alloys. Previous investigations have paid much attention on the corrosion of magnesium alloys in the presence of Cl- in atmospheric [[10], [11], [12], [13], [14]] and aqueous environments [[15], [16], [17], [18], [19], [20]]. However, the influence of NH4+ on the corrosion of magnesium alloys is underappreciated. Volovitch et al. [21] compared the corrosion mechanisms of Zn (Mg, Al) coated steel in accelerated tests and field exposure and suggested that the solution containing NH4+ exhibited a preponderant correlation between the laboratory simulation and natural exposure test. Battocchi et al. [22] also built the NH4+-containing simulated solution and demonstrated the higher corrosion rate of pure Mg in this solution as compared to that in the pure NaCl. Cao et al. [10] confirmed that NH4+ could dissolve larger amount of MgO and Mg(OH)2 than the Cl-. All of the above studies suggest that NH4+ exhibits different effects on corrosion of magnesium from Cl- and the intrinsic mechanism of NH4+ should be clarified.
The effect of NH4+ on the corrosion of metallic materials can be summarized as the following three aspects: complexation with metallic ions, modification of solution, and disruption of corrosion products. Firstly, it has been found that the corrosion of Cu and Zn was accelerated in the presence of NH4+, attributed to the formation of soluble complex compounds [[23], [24], [25]]. Secondly, the solution chemistry is modified in the presence of NH4+, of which the concentration is pH-dependent [26]. Moreover, the buffering effect of NH4+ can provide H+ and support the cathodic reactions. Cui et al. [27] found that the buffering effect of NH4+ combined with the effect of NO3- and Cl- resulted in the “autocatalytic” pitting corrosion of AZ31 magnesium alloy. Thirdly, the corrosion products can be significantly deteriorated by NH4+. Buggio et al. [28] reported that metastability of hydride phases was favored in the presence of NH4+ which could alter the equilibrium dissociation of water. Citterio et al. [29] declared that the dissociation of NH4+ into NH3 and H+ hindered the precipitation of Mg(OH)2. Cao et al. [30] proposed that NH4+ could dissolve the relatively protective MgO layer and provide a path for the aggressive electrolytes to diffuse to the substrate.
In this study, the effect of NH4+ on the corrosion behavior of AZ31 magnesium alloy is investigated systematically in sulfate solution. The corrosion rate is quantified by weight loss, hydrogen collection, and electrochemical tests. The corrosion product morphology and the surface morphology beneath the product layer are observed. The influence of NH4+ on the corrosion rate, electrochemical behavior, corrosion product formation, and surface morphologies are discussed.
2. Experimental
2.1. Materials and solution
The testing specimens were prepared from a rolled sheet with a thickness of 3.2 mm. Table 1 gives the chemical composition of AZ31 magnesium alloy used in the present work. Samples for immersion test with dimension of 20 mm × 20 mm × 3 mm were ground down to 1500 grit SiC paper. The electrochemical measurement samples with the size of 10 mm × 10 mm × 3 mm were mounted in epoxy resin, leaving 1 cm2 as the testing surface. After the grinding process, all the samples were rinsed with distilled water to remove the surface residues, degreased in anhydrous ethanol and finally dried in cold air. Then the specimens were immediately exposed to the electrolytes to conduct immersion and electrochemical tests.
Table 1
Table 1Chemical composition (wt.%) of AZ31 magnesium alloy used in the present work.
The testing solutions were 0.0005 M, 0.005 M, and 0.05 M ammonium sulfate ((NH4)2SO4), which were prepared with analytical grade reagents and deionized water. These solutions were denoted as 0.001 M, 0.01 M, and 0.1 M NH4+, respectively in the following descriptions, for both a concise presentation and an emphasis of the research point. The solution pH was measured but it was not controlled during the corrosion test. All the experiments were carried out at room temperature (25 ± 1 °C).
2.2. Immersion test
The original weight (w0) of the samples and pH of the solutions were measured before the immersion tests. The ratio of the solution volume to the sample surface area was 30 mL/cm2. Different immersion intervals (6 h, 24 h, 48 h, 72 h, 96 h, and 120 h) were designed to obtain the corrosion kinetics in different environments. After immersion test, the corroded samples were cleaned with chromate solution (200 g L-1 CrO3 + 10 g L-1 AgNO3 + 20 g L-1 BaNO3) to remove corrosion products. Finally, the corroded samples were dried in air and reweighed, documented as wn. Five parallel experiments were taken for each group at least for obtaining accurate experimental data to check the reproducibility. The corrosion rate, VW (mg cm-2 d-1), was calculated through the following equation [31]:
was the surface area of the samples, and tn (day) was the immersion time. The average corrosion rate PW (mm a-1) was calculated by Eq. (2) [32]:
${{P}_{\text{W}}}=2.10{{V}_{\text{W}}}$
After the immersion test, the surface morphologies with and without corrosion products were examined using a scanning electron microscope (SEM, Quanta 250). The corrosion products were characterized by energy dispersive X-ray spectroscopy (EDS), X-ray diffraction (XRD, Bruker D8), and Fourier transform infrared spectroscopy (FTIR, Nicolet 5700).
2.3. Hydrogen evolution
Hydrogen gas collection samples were suspended in inverted funnel filled with solution, in a beaker open to laboratory air. The evolved hydrogen gas volume was recorded regularly. The hydrogen evolution rate, VH (mL cm-2 d-1), was calculated as follows:
was hydrogen evolution volume (mL), t was the immersion time (day), n was the period of immersion. The average corrosion rate, PH (mm a-1), was evaluated through the following equation [16,33]:
${{P}_{\text{H}}}=2.279{{V}_{\text{H}}}$
2.4. Electrochemical measurements
The electrochemical measurements were performed by CHI660 electrochemical workstation at 25 ± 1 °C using the traditional three-electrode system, with the platinum plate as counter electrode, the saturated calomel electrode (SCE) as reference electrode, and the AZ31 magnesium alloy as working electrode sample. All the potentials in the following descriptions are referred to the SCE.
The open circuit potential (OCP) and solution pH were recorded simultaneously during the immersion in the electrolytes after different time intervals (0.5 h, 3 h, 6 h, 12 h, 24 h, 48 h, 72 h, 96 h, and 120 h). Then the corresponding E-pH diagrams of AZ31 magnesium alloy were depicted. Prior to the polarization curve measurement, the specimens were immersed in the solution for 10 min to obtain a relative stable state. The polarization curves were obtained from OCP to the anodic and cathodic directions separately with a scanning rate of 0.333 mV s-1. This separate scan is helpful to avoid the influence of solution variation on the anodic process. Electrochemical impedance spectroscopy (EIS) tests were conducted after stabilization for 0.5 h at OCP over a frequency ranging from 100 kHz to 10 mHz using a 10 mV amplitude sinusoidal voltage. The impedance spectra were collected during the 120 h immersion at OCP with different time intervals that were identical to the OCP tests. All the experiments were repeated at least three times to check the reproducibility.
3. Results
3.1. Weight loss and hydrogen evolution
Fig. 1 shows the weight loss (a) and corrosion rate (b) of AZ31 magnesium alloy in solutions with different concentrations of NH4+. The weight loss increases gradually with extending immersion time in the three solutions while the increasing trends differ from each other. A rough linear increase is observed in the most dilute solution, yielding a fluctuation of corrosion rate during the immersion test (Fig. 1(b), inserted image). In the solution containing 0.01 M NH4+, weight loss obeys an approximate power law function with increasing immersion time, yielding a general decrease of corrosion rate throughout the immersion test despite some deviations (Fig. 1(b), inserted image). In the most concentrated solution, the mass loss still follows a power law function during the initial 72 h immersion, after which a linear increase is detected. This results in a gradual decrease and then a sluggish increase of the corrosion rate in this environment (Fig. 1(b)). In addition, the solution pH increases gradually with increasing immersion time and stabilizes around 9.5 (Fig. S1 in Supporting Information). A prompt increase of pH during the initial immersion period because of the rapid dissolution of Mg is observed, after which the pH shows stable values. Moreover, corrosion rate of AZ31 magnesium alloy is apparently increased with increasing NH4+ concentration, with the most concentrated solution exhibiting the lowest pH.
Fig. 1.
Weight loss (a) and average corrosion rates (b) of AZ31 magnesium alloys in vary concentrations of NH4+.
Fig. 2 shows the hydrogen evolution volume (a) and the corresponding instantaneous corrosion rate (b) of AZ31 magnesium alloy during the immersion tests in solutions with different concentrations of NH4+. The released hydrogen gas volume increases gradually with extending immersion time, approximately following the power law function. The augment of hydrogen evolution volume in the final stage in the solution containing 0.001 M NH4+ is difficult to distinguish, yielding an almost constant corrosion rate that is close to 0 (Fig. 2(b)). In contrast, released hydrogen gas increases dramatically in the solution containing 0.1 M NH4+, which is not recorded after exposure for 12 h (Fig. 2(a)). In this case, the corrosion rate decreases gradually with increasing immersion time (Fig. 2(b), inserted image). Corrosion rate in the solution with 0.01 M NH4+ exhibits the similar tendency but some fluctuations are detected. The general decrease of corrosion rate is consistent with the weight loss measurement and the variation of solution pH.
Fig. 2.
Hydrogen evolution (a) and corrosion rates (b) of AZ31 magnesium alloys in the solutions with different concentrations of NH4+.
3.2. Corrosion product analysis
Fig. 3 shows the corrosion product morphologies of AZ31 magnesium alloy after immersion in the NH4+-containing solutions for different time. Corrosion is slight during the initial 6 h immersion in the solution containing 0.001 M (Fig. 3(a1)) and 0.01 M (Fig. 3(b1)) NH4+, as indicated by the clear scratches on the specimen surface. In the 0.1 M NH4+ solution, however, a distinct layer of corrosion product which is full of cracks is detected (Fig. 3(c1)). As the immersion extends to 24 h, all the samples are covered by a corrosion product layer with numerous micro-cracks. The cracks are, most likely, formed by dehydration of the corrosion layer during the drying process after immersion. Moreover, the cracks are more intensive in the dilute solution, dividing the corrosion product layer to smaller lumps. The peeling of the corrosion products occurs in the 0.01 M (Fig. 3(b3)) and 0.1 M NH4+ (Fig. 3(c2) and (c3)) solution in the later immersion stage, suggesting the poor adherence of this loose layer. The typical points marked in Fig. 3(c) are detected by EDS. The results reveal that corrosion products are mainly composed of Mg, O, C, and Al (Table S1, in Supporting Information). Obviously, the higher content of Mg in the point I and IV is attributed to the closer distance to the specimen surface and more signals coming from the matrix. The other points (III, V, and VI) have the similar elemental composition, which may be ascribed to the formation of compounds containing Mg, Al, O, and C.
Fig. 3.
SEM morphologies of the surface corrosion products on AZ31 magnesium alloy after immersion in the solutions containing 0.001 M (a1)-(a3), 0.01 M (b1)-(b3), and 0.1 M (c1)-(c3) NH4+ for 6 h (a1)-(c1), 24 h (a2)-(c2), and 120 h (a3)-(c3). (The delineated points are detected by EDS.).
Fig. 4 shows the FTIR (a) and XRD (b) spectra of the corrosion products formed on AZ31 magnesium alloy after immersion in different solutions for 120 h. The broad bands around 1640 cm-1 and 3436 cm-1 are attributed to the bending vibrations and stretching vibrations of H2O and H2O/OH- groups, respectively [34]. The peaks around 1400 cm-1 is ascribed to the presence of CO32- [35]. The peak at 1100 cm-1 and the associated peaks at lower wavenumbers can be assigned to the presence of Mg5(CO3)4(OH)2·xH2O (x = 4 or 5) [36]. The peaks in the XRD spectra are not obvious because the surface corrosion products are easy to be peeled off. Even so, the peaks centered at 11.1°, 22.2°, and 60.8° imply the existence of aluminum magnesium hydroxide carbonate hydrate ((Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O) [13]. The corrosion product powders dropped from the specimen are also collected and examined by XRD (Fig. S2, in Supporting Information), and the extra peaks indicate the presence of Mg5(CO3)4(OH)2·5H2O.
Fig. 4.
FTIR (a) and XRD (b) spectra of the corrosion products formed on AZ31 magnesium alloy surface after immersion in different solutions for 120 h.
Fig. 5 shows the cross-sectional morphology of AZ31 magnesium alloy after immersion for 120 h in solutions with different concentrations of NH4+. In 0.001 M NH4+ solution, occurrence of knife-cut like corrosion (about 17 μm in thickness) and its propagation beneath the thin corrosion product film is observed (Fig. 5(a)). This typical morphology is considered a kind of localized corrosion and occurs in the initial stage of immersion. When the NH4+ concentration is 0.01 M (Fig. 5(b) and (d)), there are still some knife-cut like cracks (∼13 μm) under the corrosion product layer (∼20 μm). In 0.1 M NH4+ solution (Fig. 5(c)), no knife-cut like crack is observed beneath the corrosion product layer (∼130 μm), attributed to the uniform corrosion. In addition, the thickness of the corrosion product layer increases with increasing NH4+ content, suggesting the more severe dissolution of Mg in concentrated solutions.
Fig. 5.
Cross-sectional morphology of corrosion product and the substrate beneath it of AZ31 magnesium alloys after 120 h immersion in solutions with 0.001 M (a), 0.01 M (b), (d), and 0.1 M NH4+ (c).
3.3. Surface morphology after removing corrosion products
In order to reveal the corrosion process under the surface layer, the surface morphology of the AZ31 magnesium alloy after removing corrosion products is observed and shown in Fig. 6. In 0.001 M NH4+ solution (Fig. 6(a)), the knife-cut like corrosion takes place after immersion for 24 h and then extends to all over the alloy surface after immersion for 120 h. In the final exposure stage, there are still some intact regions, implying the slight corrosion of the alloy in this environment (Fig. 6(a3)). However, no intact region can be observed in 0.01 M NH4+ solution (Fig. 6(b)). Instead, some trenches that may be due to the grain boundary attacks are detected (Fig. 6(b2) and (b3)). When the NH4+ concentration is high (0.1 M), the alloy shows uniform corrosion with a flat surface and some randomly distributed Al-Mn particles (Fig. 6(c)).
Fig. 6.
Surface morphologies of AZ31 magnesium alloy without corrosion products after immersion in the solutions containing 0.001 M (a1)-(a3), 0.01 M (b1)-(b3), and 0.1 M (c1)-(c3) NH4+ for 6 h (a1)-(c1), 24 h (a2)-(c2), and 120 h (a3)-(c3).
3.4. Electrochemical measurement
3.4.1. Open circuit potential and solution pH
Fig. 7 shows the time-dependence of OCP (a) and solution pH (b) of AZ31 magnesium alloy during immersion in different solutions, as well as the corresponding E-pH diagram of this system (c) and the theoretical overlaid E-pH diagram of N-H2O and Mg-H2O system (d). The OCPs increase sharply in the initial immersion stage and then tend to stabilize with further immersion. The steady state OCP is shifted negatively with increasing NH4+ concentration (Fig. 7(a)). The pH increase tendency is similar in different solutions but the final pH is lower in the dilute solution than that in concentrated ones (Fig. 7(b)). The sharp increase in the initial immersion period corresponds well with the quick increase of OCP. When comparing the E-pH diagram of this system and the overlaid N-H2O and Mg-H2O system, active dissolution of AZ31 magnesium alloys can be predicted as indicated by the steady presence of Mg2+ (Fig. 7(d)).
Fig. 7.
Changes of OCPs (a) and pH values (b) of AZ31 magnesium alloy in NH4+ solutions and the corresponding E-pH diagram (c), as well as the overlaid E-pH diagram of the N-H2O and Mg-H2O system (d).
3.4.2. Polarization curve
Fig. 8 shows the polarization curves of the AZ31 magnesium alloy in solutions with different concentrations of NH4+ before (a) and after (b) IR compensation. The data processing of IR correction is to compensate the influence of solution resistance on the potential drop because the disparities of this parameter (obtained by EIS) among the three solutions are considerable enough to affect the polarization curves [37]. The corrosion potentials (Ecorr), cathodic Tafel slope (bc), and the corrosion current density (icorr) are extrapolated from the polarization curves and listed in Table 2. It should be noted that only cathodic polarization curves are used to conduct the Tafel fitting because the anodic part does not meet the strict prerequisites of Tafel fitting. Among them, the Ecorr corresponds to short-term OCP because the curves are scanned from OCP to positive and negative directions separately. Both the anodic and cathodic reactions are expedited with increasing NH4+ concentration, yielding an overall curve shift to the lower right direction. This also leads to the negative shift of the Ecorr from -1.524 to -1.722 VSCE, which is consistent with the long-term OCP evolutions (Fig. 7(a)). Moreover, the corrosion current density increases almost 70 times as compared the values in the 0.001 M and 0.1 M NH4+ solutions (Table 2).
Fig. 8.
Polarization curves of AZ31 magnesium alloy in solutions with different concentrations of NH4+: (a) before IR compensation, (b) after IR compensation.
Table 2
Table 2Corrosion parameters of AZ31 magnesium alloys in different (NH4)2SO4 solution derived from the IR-compensated polarization curves.
Fig. 9 shows the EIS of AZ31 magnesium alloy in solutions with different concentrations of NH4+ during the immersion tests. As shown in Fig. 9(a), the Nyquist spectra of the specimens in 0.001 M NH4+ solution are consisted of two capacitive loops at the high- and low-frequency range, respectively. As the NH4+ concentration is increased to 0.01 M, the Nyquist spectra are composed of two capacitive loops and a low-frequency inductive loop. When the NH4+ concentration is increased to 0.1 M, there are two capacitive loops and two inductive loops at the initial immersion stage. But the low-frequency inductive loop disappears with extending immersion time. The diameter of the capacitive loops in 0.001 M NH4+ solution fluctuates with increasing immersion time while that in the other two electrolytes increases continuously as the immersion test extends.
Fig. 9.
Nyquist diagrams and fitting lines of AZ31 magnesium alloys after immersion for different time in the solutions containing 0.001 M (a), 0.01 M (b), and 0.1 M NH4+ (c).
Corresponding equivalent circuits used for analysis of the data measured in NH4+ solutions after different immersion periods are shown in Fig. 10(a). In the equivalent circuits, Rs is solution resistance, Rct is the charge transfer resistance, CPEdl is the double layer capacitance, Rf and CPEf represent the corrosion product film resistance and its capacitance, L1 and L2 are inductance and RL1 and RL2 are the corresponding resistance, of which the physical meaning will be analyzed in the discussion section. The extrapolated parameters of the AZ31 magnesium alloy after fitting with the equivalent circuits are listed in Table 3. The polarization resistance (Rp) in different models can be expressed by the following equations:
Fig. 10(b) shows the variations of Rp during the immersion test. A rough increase with some fluctuations is observed in 0.001 M and 0.01 M NH4+ solutions, while Rp in 0.1 M NH4+ solutions shows a monotonous increase with extending immersion time. The Rp can be transferred to corrosion current density using the Stern equation with the constant B of 60 mV [38]. Then the corrosion rate Peis (mm a-1) can be calculated through the following equation and will be discussed in the following section [31,39,40]:
${{P}_{\text{eis}}}=22.85{{i}_{\text{corr}}}$
4. Discussion
4.1. Effect of NH4+ on the corrosion rate of AZ31 magnesium alloy
Fig. 11 compares the corrosion rate obtained by weight loss, hydrogen evolution, and EIS after immersion for different time. A general decrease of corrosion rate in the three solutions is observed despite some fluctuations in dilute environments. The decrease of corrosion rate with increasing immersion can be ascribed to the increase of solution pH [41,42], formation of corrosion products which decreases the active surface area [[43], [44], [45]], and the decrease of NH4+ content at high pH environment [27]. The fluctuation of corrosion rate in solution with 0.001 M NH4+ is attributed to the local rupture of the surface film [10,41].
Fig. 11.
Comparison of the corrosion rates of AZ31 magnesium alloy during immersion in the NH4+-containing solutions for different time. (b) is the local magnification image marked in (a).
Notably, there exist dissimilarities between the corrosion rates obtained by different methods because of the intrinsic differences of these approaches. Generally, the corrosion rate calculated by hydrogen evolution is relatively lower as compared to other techniques. On one hand, the increase in the released hydrogen volume is low in the final stage and is difficult to differentiate. On the other hand, formation of hydride sub-products consumes the generated H2 and decreases the apparent corrosion rate [5,28,46]. Buggio et al. [28] revealed that the chemical recombination of hydrogen atoms of oxidic metastable hydrides was the kinetically determinant process during anodic oxidation of Mg in NH4+-containing electrolytes. In the 0.001 M NH4+ solution, corrosion rate derived from weight loss method is the highest, which may be attributed to the Non-Faraday process, including the undermining of second phases [47] and the dissolution of univalent magnesium ions [33], that cannot be detected by electrochemical methods. In 0.01 M and 0.1 M NH4+ solution, corrosion rate obtained by EIS is higher than that obtained by weight loss, especially in the 0.1 M NH4+ solution. Several reasons may be responsible for this phenomenon. Firstly, the electrochemical dissolution process may overshadow the Non-Faraday process, yielding a higher corrosion rate obtained by EIS. Secondly, EIS is an instantaneous approach, which provides a corrosion rate at a particular time over which the test is performed [8]. Therefore, this rate may be higher than the average corrosion rate that extracted from mass-loss method.
4.2. Effect of NH4+ on corrosion products and morphologies of AZ31 magnesium alloy
The original pH values of the solutions containing 0.001 M, 0.01 M, and 0.1 M NH4+ are 6.13, 5.97, and 5.62, respectively. This is attributed to the hydrolysis of ammonium which generates H+ as follows:
Cui et al. [27] reported that the amount of NH4+ is more than 90% in a solution with a pH less than 8. Therefore, NH4+ can influence the whole corrosion process of AZ31 magnesium alloy. Firstly, the complexation of NH4+ is excluded because the complexing action between NH4+ and Mg is negligible and the Zn in AZ31 alloy is limited to cause a considerable complexation [27,48]. Even so, the presence of NH4+ may affect the surface corrosion products and expedite the corrosion processes. In Fig. 1(c), the pH values of all solutions after a 5-day immersion are below 8.4, under which the stable Mg(OH)2 layer is difficult to be preserved because the precipitation of brucite is possible when the pH exceeds 9.43 [49]. This has been confirmed by XRD and FTIR results (Fig. 4) where the peak from Mg(OH)2 is not detected. Additionally, Cao et al. [30] reported that the acceleration effect of NH4+ on Mg corrosion could be attributed to the dissolution of the thin protective inner MgO layer/outer non-protective Mg(OH)2 layer in the corrosion product film. The dissolution reactions are as follows:
Moreover, ammonium ion preferentially degrades MgO, which is assumed to be more protective than Mg(OH)2 [30,43,50]. Actually, there must be a thin Mg(OH)2 and MgO film formed at the interface of solution and magnesium, which may be caused by the partial alkalization on account of the cathodic reactions during dissolution of magnesium [51]. Due to the dissolution of the corrosion product layer, a fresh surface of AZ31 magnesium alloy is exposed, and the substrate can directly react with the solution without any barrier. Eventually, the active surface area of AZ31 magnesium alloy decreases and the bare surface is covered by a loose corrosion product film, which may be constituted by various ions through a weak-interaction.
XRD analysis implies the presence of Mg-Al-CO32- hydrotalcite-like compound (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O (HTLc) and Mg5(CO3)4(OH)2·5H2O (Section 3.2). The presence of Mg5(CO3)4(OH)2·5H2O is likely attributed to the diffusion of CO2 from the environment, which reacts with unstable Mg(OH)2 [52,53]. Arrabal et al. [54,55] found the presence of (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O on the magnesium alloy after immersion in 3.5 wt.% NaCl. Jang et al. [56] also found that the (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O was the primary corrosion product on AZ31 alloy in simulated physiological solutions. In atmospheric environment, Liao et al. [13] demonstrated the existence of the HTLc as the dominant corrosion products and proposed a simplified formation process. They considered that the high temperature on sunny days and the high local pH promoted the dissolution of Al3+ ions and thus the (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O was developed. In the present work, the solution is acidic in the initial immersion period in the presence of NH4+. This could accelerate the dissolution of Al to Al3+ and enhance the formation of (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O in the later immersion stage, during which the formation of Al-rich precursors of hydrotalcite (HT) and exchange of OH- for CO32- are feasible [57]. This HT-like compound is similar to HT and presents a structure related to brucite with the general formula [Mg1-x2+Mx3+(OH)2]x+Ax/nn-·mH2O, where M2+ and M3+ are divalent and trivalent metals, respectively, and An- is an anion [13,57].
As shown in Fig. 5, Fig. 6, the surface morphologies beneath the corrosion products change greatly with increasing NH4+ content. A transformation from knife-cut like localized corrosion to uniform corrosion occurs, which implies the accelerating effect of NH4+. Fig. 12 shows the schematic diagram of the corrosion process of AZ31 magnesium alloy in NH4+-containing solutions. In 0.001 M and 0.01 M NH4+ solution, knife-cut like corrosion penetrates deeper in some active areas of the alloy surface, and the presence of NH4+ ions in these active points can significantly delay the formation of MgO, resulting in the knife-cut like corrosion. Moreover, due to the dissolution of corrosion film (MgO) by NH4+, there is no protective film on the surface, which causes the uniform corrosion of AZ31 magnesium in 0.1 M NH4+ solution. The corrosion acceleration effect caused by NH4+ is higher in the concentrated NH4+ solutions.
Fig. 12.
Schematic diagram of the corrosion process of AZ31 magnesium alloy in NH4+-containing solutions.
4.3. Effect of NH4+ on the electrochemical behavior of magnesium alloy
4.3.1. Polarization curve analysis
According to Fig. 8(a), both anodic and cathodic reaction kinetics of AZ31 magnesium alloy are promoted with increasing NH4+ concentration. It is well known that the cathodic process of Mg in aqueous solution is mainly hydrogen evolution process with the following reaction:
The cathodic current associated with the reduction of oxygen can be neglected since the oxygen reduction is not important to Mg corrosion [44]. At the initial immersion stage, hydrolysis of NH4+ results in an acidic environment and promotes the cathodic process. In NH4+-containing solution, the anodic process is very complex. The reactions are as follows:
At the initial immersion stage, reactions (13) and (15) are the main reaction of the anodic process. However, reaction (14) will gradually replace reaction (13) and Mg(OH)2 film will form with extending immersion time because of the partial alkalization. In this case, NH4+ has two main influences: (i) hydrolysis of NH4+ could produce H+ and consumes OH-, promoting reactions (14) and (16); (ii) NH4+ will dissolve the corrosion film and expose the Mg surface, facilitating reactions (14) and (15). Therefore, the anodic process can be promoted extremely. There is no breaking potential (Ebp) at the anodic branch of polarization curves in all (NH4)2SO4 solutions, indicating the rapid corrosion rate of AZ31 in NH4+-containing solution and weak protection of the corrosion products.
As reported by Buggio et al. [28], the microstructure of Mg alloy, the nature of the second salt or its proportion appeared to be unimportant as the anodic (or cathodic) potential was raised in NH4+-based solution. Pébère et al. [45] investigated the electrochemical behavior of magnesium using SO42- as a supporting electrolyte because of the low aggressive of SO42-. Cui et al. [58] reported that SO42- ions had no obvious effect on the icorr in saline solution. Zeng et al. [42] reported that SO42- exhibited little effect on the corrosion of magnesium alloy. Therefore, compared with the strong effect of NH4+ ion, the effect of SO42- on Mg corrosion is negligible. Potentiodynamic polarization curves of AZ31 magnesium in 0.005 M (NH4)2SO4 and Na2SO4 (pH values adjusted with H2SO4) are shown in Fig. 13 and the extracted parameters are listed in Table 4. As shown in Fig. 13, the cathodic Tafel slope of the polarization curve increases with the addition of NH4+. Moreover, there is a pseudo-passive region at the anodic branch of AZ31 in all Na2SO4 solutions. Therefore, Fig. 13 indicates that the protective MgO film of the surface could be dissolved by the NH4+. However, the influence of H+ on the corrosion behavior of AZ31 magnesium can be neglected. In view of this concentration, the effect of NH4+ on the corrosion rate of the AZ31 magnesium alloy is over 88%. Similar results are also found when comparing the polarization curves in 0.05 M (NH4)2SO4 and Na2SO4 with the same original pH (Fig. S3, in Supporting Information).
The interfacial process occurring on the magnesium/solution interface varies with NH4+ concentration as indicated by the change of EIS loops. To intuitively present the evolution of EIS data with increasing immersion time and NH4+ concentration, the normalized impedance diagrams which are proposed by Baril et al. [43] are plotted and shown in Fig. 14. The diagrams measured in 0.001 M NH4+ solution coincide well with each other, indicating the same corrosion mechanism during the immersion test [59]. In 0.01 M NH4+ solution, the inductive loops shrink with increasing immersion time, while an additional inductive loop is detected at low frequency in the 0.1 M NH4+ solution, suggesting that aging in the solution affects the interfacial processes. Moreover, the diagrams at 0.5 h in the three solutions show distinct characteristics, revealing that NH4+ concentration also influence the dissolution process occurring on the magnesium/solution interface.
Fig. 14.
Normalized impedance diagrams of AZ31 magnesium alloys after immersion in the solutions containing 0.001 M (a), 0.01 M (b), and 0.1 M (c) NH4+ for different time and the comparison of the diagrams in different solutions at 0.5 h (d).
Firstly, the two capacitive loops at high frequency and low frequency in all the three solutions can be attributed to the charge transfer process and the corrosion product film, respectively. Both of the charge transfer resistance and film resistance significantly decrease with increasing NH4+ concentration, suggesting the acceleration of the corrosion process. According to Cao et al. [30], the inner MgO layer was easily to be dissolved by NH4+ as compared to the Mg(OH)2. This creates more active surface area and results in the expedited corrosion process in the high NH4+ solutions [43,44]. With increasing immersion time, the Rct and Rf increase gradually in the 0.01 M and 0.1 M NH4+ solutions, revealing the blocking effect of the corrosion products that precipitated on the sample surface. The fluctuations of Rct and Rf in 0.001 M NH4+ solution can be ascribed to the competition between film growth and dissolution processes [41,46,60,61] on account of the slight corrosion in this environment. In addition, the variation of the capacitance values may be attributed to the variation of the film thickness and the available area.
Secondly, inductive loops appear in the EIS diagrams of AZ31 magnesium alloys in 0.01 M and 0.1 M NH4+ solutions, with the latter exhibiting two loops during the initial immersion periods. Several mechanisms have been proposed to interpret the inductance loops in EIS spectra of magnesium alloys in the literatures [30,37,59,[62], [63], [64], [65]], including the Mg+ theory, the adsorption/desorption of intermediate species, and the initiation of localized corrosion. As shown in Fig. 3, the distinct difference between the surface layer morphology in 0.001 M NH4+ and the other solutions is the relatively intact surface film without obvious spalling (Fig. 3(a2) and (a3)). Therefore, the inductive loops in 0.01 M and 0.1 M NH4+ may be attributed to the local destruction of surface layer which contributes to the occurrence of localized corrosion and the adsorption of intermediate species. This agrees well with Gomes et al. [59] and Chen et al. [66] who found the similar inductive loops in the sulfate-based solutions. The other inductive loop in the lowest frequency range in 0.1 M NH4+ solution may be attributed to the adsorption of hydrogen atoms. In this electrolyte, the prominent characteristic is the severe hydrogen evolution during the initial immersion period (Fig. 2(a)). Because hydrogen evolution reaction can be divided into two procedures including the electrochemical dissolution and electrochemical desorption, the lower rate of the latter one creates considerable Hads on the sample surface. According to Cao et al. [67] and Armstrong et al. [68], this intermediate product yields an inductive loop which can reasonably explains the particular inductive loop in the most concentrated solution during the initial immersion stage.
5. Conclusions
In this study, the corrosion behavior of AZ31 magnesium alloy in the sulfate solutions with different concentrations of NH4+ was investigated. The conclusion can be summarized as follows:
(1) Corrosion rate of AZ31 magnesium increases with the addition of (NH4)2SO4 because of the degradation of the surface product layer by NH4+.
(2) With the increase of NH4+ content, the surface morphologies beneath the corrosion products change from knife-cut like localized corrosion to uniform corrosion.
(3) Corrosion products are mainly composed of (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O and Mg5(CO3)4(OH)2·5H2O, which is independent of the NH4+ concentration.
(4) Both cathodic and anodic reactions are accelerated by NH4+ ions but the promotion of the anodic reactions is more obvious, resulting in the negative shift of the free corrosion potential.
(5) Interfacial proesses occurring on magnesium/solution interface changes with NH4+ concentration and immersion time. The occurrence of localized corrosion and severe evolution of hydrogen contribute to the inductive loops.
Acknowledgements
The authors wish to acknowledgement the financial support of Science and Technology Support Plan for Youth Innovation of Colleges in Shandong Province (No. DC2000000891) and the National Natural Science Foundation of China (Nos. 51701102 and 51601182).
Rapid industrialization and urbanization in developing countries has led to an increase in air pollution, along a similar trajectory to that previously experienced by the developed nations. In China, particulate pollution is a serious environmental problem that is influencing air quality, regional and global climates, and human health. In response to the extremely severe and persistent haze pollution experienced by about 800 million people during the first quarter of 2013 (refs 4, 5), the Chinese State Council announced its aim to reduce concentrations of PM2.5 (particulate matter with an aerodynamic diameter less than 2.5 micrometres) by up to 25 per cent relative to 2012 levels by 2017 (ref. 6). Such efforts however require elucidation of the factors governing the abundance and composition of PM2.5, which remain poorly constrained in China. Here we combine a comprehensive set of novel and state-of-the-art offline analytical approaches and statistical techniques to investigate the chemical nature and sources of particulate matter at urban locations in Beijing, Shanghai, Guangzhou and Xi'an during January 2013. We find that the severe haze pollution event was driven to a large extent by secondary aerosol formation, which contributed 30-77 per cent and 44-71 per cent (average for all four cities) of PM2.5 and of organic aerosol, respectively. On average, the contribution of secondary organic aerosol (SOA) and secondary inorganic aerosol (SIA) are found to be of similar importance (SOA/SIA ratios range from 0.6 to 1.4). Our results suggest that, in addition to mitigating primary particulate emissions, reducing the emissions of secondary aerosol precursors from, for example, fossil fuel combustion and biomass burning is likely to be important for controlling China's PM2.5 levels and for reducing the environmental, economic and health impacts resulting from particulate pollution.
... For decades, the rapid development of industry has aggravated air pollution in several developing countries [[1], [2], [3]]. The air pollution not only does harm to human health, but also affects the corrosion of some metallic materials because of the presence of particulate matter (PM). It is well-known that water-soluble inorganic ions (WSIIs) are important ingredients in PM2.5 mass. Zhang et al. [4] reported that the main WSIIs are sulfate (SO42-), ammonium (NH4+), chloride (Cl-), and nitrate ions (NO3-). When pollution particles containing these ions come into contact with the metal surface, they can easily absorb water from the surroundings and form a thin electrolyte layer covering the metal surface [5]. This creates an aggressive environment that attacks metallic materials. Therefore, it is meaningful to study the influence of these ions on the corrosion of metallic materials. ...
1
2016
... For decades, the rapid development of industry has aggravated air pollution in several developing countries [[1], [2], [3]]. The air pollution not only does harm to human health, but also affects the corrosion of some metallic materials because of the presence of particulate matter (PM). It is well-known that water-soluble inorganic ions (WSIIs) are important ingredients in PM2.5 mass. Zhang et al. [4] reported that the main WSIIs are sulfate (SO42-), ammonium (NH4+), chloride (Cl-), and nitrate ions (NO3-). When pollution particles containing these ions come into contact with the metal surface, they can easily absorb water from the surroundings and form a thin electrolyte layer covering the metal surface [5]. This creates an aggressive environment that attacks metallic materials. Therefore, it is meaningful to study the influence of these ions on the corrosion of metallic materials. ...
1
2018
... For decades, the rapid development of industry has aggravated air pollution in several developing countries [[1], [2], [3]]. The air pollution not only does harm to human health, but also affects the corrosion of some metallic materials because of the presence of particulate matter (PM). It is well-known that water-soluble inorganic ions (WSIIs) are important ingredients in PM2.5 mass. Zhang et al. [4] reported that the main WSIIs are sulfate (SO42-), ammonium (NH4+), chloride (Cl-), and nitrate ions (NO3-). When pollution particles containing these ions come into contact with the metal surface, they can easily absorb water from the surroundings and form a thin electrolyte layer covering the metal surface [5]. This creates an aggressive environment that attacks metallic materials. Therefore, it is meaningful to study the influence of these ions on the corrosion of metallic materials. ...
1
2012
... For decades, the rapid development of industry has aggravated air pollution in several developing countries [[1], [2], [3]]. The air pollution not only does harm to human health, but also affects the corrosion of some metallic materials because of the presence of particulate matter (PM). It is well-known that water-soluble inorganic ions (WSIIs) are important ingredients in PM2.5 mass. Zhang et al. [4] reported that the main WSIIs are sulfate (SO42-), ammonium (NH4+), chloride (Cl-), and nitrate ions (NO3-). When pollution particles containing these ions come into contact with the metal surface, they can easily absorb water from the surroundings and form a thin electrolyte layer covering the metal surface [5]. This creates an aggressive environment that attacks metallic materials. Therefore, it is meaningful to study the influence of these ions on the corrosion of metallic materials. ...
2
2008
... For decades, the rapid development of industry has aggravated air pollution in several developing countries [[1], [2], [3]]. The air pollution not only does harm to human health, but also affects the corrosion of some metallic materials because of the presence of particulate matter (PM). It is well-known that water-soluble inorganic ions (WSIIs) are important ingredients in PM2.5 mass. Zhang et al. [4] reported that the main WSIIs are sulfate (SO42-), ammonium (NH4+), chloride (Cl-), and nitrate ions (NO3-). When pollution particles containing these ions come into contact with the metal surface, they can easily absorb water from the surroundings and form a thin electrolyte layer covering the metal surface [5]. This creates an aggressive environment that attacks metallic materials. Therefore, it is meaningful to study the influence of these ions on the corrosion of metallic materials. ...
... Notably, there exist dissimilarities between the corrosion rates obtained by different methods because of the intrinsic differences of these approaches. Generally, the corrosion rate calculated by hydrogen evolution is relatively lower as compared to other techniques. On one hand, the increase in the released hydrogen volume is low in the final stage and is difficult to differentiate. On the other hand, formation of hydride sub-products consumes the generated H2 and decreases the apparent corrosion rate [5,28,46]. Buggio et al. [28] revealed that the chemical recombination of hydrogen atoms of oxidic metastable hydrides was the kinetically determinant process during anodic oxidation of Mg in NH4+-containing electrolytes. In the 0.001 M NH4+ solution, corrosion rate derived from weight loss method is the highest, which may be attributed to the Non-Faraday process, including the undermining of second phases [47] and the dissolution of univalent magnesium ions [33], that cannot be detected by electrochemical methods. In 0.01 M and 0.1 M NH4+ solution, corrosion rate obtained by EIS is higher than that obtained by weight loss, especially in the 0.1 M NH4+ solution. Several reasons may be responsible for this phenomenon. Firstly, the electrochemical dissolution process may overshadow the Non-Faraday process, yielding a higher corrosion rate obtained by EIS. Secondly, EIS is an instantaneous approach, which provides a corrosion rate at a particular time over which the test is performed [8]. Therefore, this rate may be higher than the average corrosion rate that extracted from mass-loss method. ...
1
2018
... Magnesium (Mg) alloys including AZ and AM series are widely used in automotive, aircraft, electronic equipment, and other lightweight fields [[6], [7], [8], [9]]. However, the poor corrosion resistance is the most important property that limits the application of these alloys. Previous investigations have paid much attention on the corrosion of magnesium alloys in the presence of Cl- in atmospheric [[10], [11], [12], [13], [14]] and aqueous environments [[15], [16], [17], [18], [19], [20]]. However, the influence of NH4+ on the corrosion of magnesium alloys is underappreciated. Volovitch et al. [21] compared the corrosion mechanisms of Zn (Mg, Al) coated steel in accelerated tests and field exposure and suggested that the solution containing NH4+ exhibited a preponderant correlation between the laboratory simulation and natural exposure test. Battocchi et al. [22] also built the NH4+-containing simulated solution and demonstrated the higher corrosion rate of pure Mg in this solution as compared to that in the pure NaCl. Cao et al. [10] confirmed that NH4+ could dissolve larger amount of MgO and Mg(OH)2 than the Cl-. All of the above studies suggest that NH4+ exhibits different effects on corrosion of magnesium from Cl- and the intrinsic mechanism of NH4+ should be clarified. ...
1
2016
... Magnesium (Mg) alloys including AZ and AM series are widely used in automotive, aircraft, electronic equipment, and other lightweight fields [[6], [7], [8], [9]]. However, the poor corrosion resistance is the most important property that limits the application of these alloys. Previous investigations have paid much attention on the corrosion of magnesium alloys in the presence of Cl- in atmospheric [[10], [11], [12], [13], [14]] and aqueous environments [[15], [16], [17], [18], [19], [20]]. However, the influence of NH4+ on the corrosion of magnesium alloys is underappreciated. Volovitch et al. [21] compared the corrosion mechanisms of Zn (Mg, Al) coated steel in accelerated tests and field exposure and suggested that the solution containing NH4+ exhibited a preponderant correlation between the laboratory simulation and natural exposure test. Battocchi et al. [22] also built the NH4+-containing simulated solution and demonstrated the higher corrosion rate of pure Mg in this solution as compared to that in the pure NaCl. Cao et al. [10] confirmed that NH4+ could dissolve larger amount of MgO and Mg(OH)2 than the Cl-. All of the above studies suggest that NH4+ exhibits different effects on corrosion of magnesium from Cl- and the intrinsic mechanism of NH4+ should be clarified. ...
2
2019
... Magnesium (Mg) alloys including AZ and AM series are widely used in automotive, aircraft, electronic equipment, and other lightweight fields [[6], [7], [8], [9]]. However, the poor corrosion resistance is the most important property that limits the application of these alloys. Previous investigations have paid much attention on the corrosion of magnesium alloys in the presence of Cl- in atmospheric [[10], [11], [12], [13], [14]] and aqueous environments [[15], [16], [17], [18], [19], [20]]. However, the influence of NH4+ on the corrosion of magnesium alloys is underappreciated. Volovitch et al. [21] compared the corrosion mechanisms of Zn (Mg, Al) coated steel in accelerated tests and field exposure and suggested that the solution containing NH4+ exhibited a preponderant correlation between the laboratory simulation and natural exposure test. Battocchi et al. [22] also built the NH4+-containing simulated solution and demonstrated the higher corrosion rate of pure Mg in this solution as compared to that in the pure NaCl. Cao et al. [10] confirmed that NH4+ could dissolve larger amount of MgO and Mg(OH)2 than the Cl-. All of the above studies suggest that NH4+ exhibits different effects on corrosion of magnesium from Cl- and the intrinsic mechanism of NH4+ should be clarified. ...
... Notably, there exist dissimilarities between the corrosion rates obtained by different methods because of the intrinsic differences of these approaches. Generally, the corrosion rate calculated by hydrogen evolution is relatively lower as compared to other techniques. On one hand, the increase in the released hydrogen volume is low in the final stage and is difficult to differentiate. On the other hand, formation of hydride sub-products consumes the generated H2 and decreases the apparent corrosion rate [5,28,46]. Buggio et al. [28] revealed that the chemical recombination of hydrogen atoms of oxidic metastable hydrides was the kinetically determinant process during anodic oxidation of Mg in NH4+-containing electrolytes. In the 0.001 M NH4+ solution, corrosion rate derived from weight loss method is the highest, which may be attributed to the Non-Faraday process, including the undermining of second phases [47] and the dissolution of univalent magnesium ions [33], that cannot be detected by electrochemical methods. In 0.01 M and 0.1 M NH4+ solution, corrosion rate obtained by EIS is higher than that obtained by weight loss, especially in the 0.1 M NH4+ solution. Several reasons may be responsible for this phenomenon. Firstly, the electrochemical dissolution process may overshadow the Non-Faraday process, yielding a higher corrosion rate obtained by EIS. Secondly, EIS is an instantaneous approach, which provides a corrosion rate at a particular time over which the test is performed [8]. Therefore, this rate may be higher than the average corrosion rate that extracted from mass-loss method. ...
1
2008
... Magnesium (Mg) alloys including AZ and AM series are widely used in automotive, aircraft, electronic equipment, and other lightweight fields [[6], [7], [8], [9]]. However, the poor corrosion resistance is the most important property that limits the application of these alloys. Previous investigations have paid much attention on the corrosion of magnesium alloys in the presence of Cl- in atmospheric [[10], [11], [12], [13], [14]] and aqueous environments [[15], [16], [17], [18], [19], [20]]. However, the influence of NH4+ on the corrosion of magnesium alloys is underappreciated. Volovitch et al. [21] compared the corrosion mechanisms of Zn (Mg, Al) coated steel in accelerated tests and field exposure and suggested that the solution containing NH4+ exhibited a preponderant correlation between the laboratory simulation and natural exposure test. Battocchi et al. [22] also built the NH4+-containing simulated solution and demonstrated the higher corrosion rate of pure Mg in this solution as compared to that in the pure NaCl. Cao et al. [10] confirmed that NH4+ could dissolve larger amount of MgO and Mg(OH)2 than the Cl-. All of the above studies suggest that NH4+ exhibits different effects on corrosion of magnesium from Cl- and the intrinsic mechanism of NH4+ should be clarified. ...
3
2013
... Magnesium (Mg) alloys including AZ and AM series are widely used in automotive, aircraft, electronic equipment, and other lightweight fields [[6], [7], [8], [9]]. However, the poor corrosion resistance is the most important property that limits the application of these alloys. Previous investigations have paid much attention on the corrosion of magnesium alloys in the presence of Cl- in atmospheric [[10], [11], [12], [13], [14]] and aqueous environments [[15], [16], [17], [18], [19], [20]]. However, the influence of NH4+ on the corrosion of magnesium alloys is underappreciated. Volovitch et al. [21] compared the corrosion mechanisms of Zn (Mg, Al) coated steel in accelerated tests and field exposure and suggested that the solution containing NH4+ exhibited a preponderant correlation between the laboratory simulation and natural exposure test. Battocchi et al. [22] also built the NH4+-containing simulated solution and demonstrated the higher corrosion rate of pure Mg in this solution as compared to that in the pure NaCl. Cao et al. [10] confirmed that NH4+ could dissolve larger amount of MgO and Mg(OH)2 than the Cl-. All of the above studies suggest that NH4+ exhibits different effects on corrosion of magnesium from Cl- and the intrinsic mechanism of NH4+ should be clarified. ...
... -containing simulated solution and demonstrated the higher corrosion rate of pure Mg in this solution as compared to that in the pure NaCl. Cao et al. [10] confirmed that NH4+ could dissolve larger amount of MgO and Mg(OH)2 than the Cl-. All of the above studies suggest that NH4+ exhibits different effects on corrosion of magnesium from Cl- and the intrinsic mechanism of NH4+ should be clarified. ...
... Fig. 11 compares the corrosion rate obtained by weight loss, hydrogen evolution, and EIS after immersion for different time. A general decrease of corrosion rate in the three solutions is observed despite some fluctuations in dilute environments. The decrease of corrosion rate with increasing immersion can be ascribed to the increase of solution pH [41,42], formation of corrosion products which decreases the active surface area [[43], [44], [45]], and the decrease of NH4+ content at high pH environment [27]. The fluctuation of corrosion rate in solution with 0.001 M NH4+ is attributed to the local rupture of the surface film [10,41]. ...
1
2014
... Magnesium (Mg) alloys including AZ and AM series are widely used in automotive, aircraft, electronic equipment, and other lightweight fields [[6], [7], [8], [9]]. However, the poor corrosion resistance is the most important property that limits the application of these alloys. Previous investigations have paid much attention on the corrosion of magnesium alloys in the presence of Cl- in atmospheric [[10], [11], [12], [13], [14]] and aqueous environments [[15], [16], [17], [18], [19], [20]]. However, the influence of NH4+ on the corrosion of magnesium alloys is underappreciated. Volovitch et al. [21] compared the corrosion mechanisms of Zn (Mg, Al) coated steel in accelerated tests and field exposure and suggested that the solution containing NH4+ exhibited a preponderant correlation between the laboratory simulation and natural exposure test. Battocchi et al. [22] also built the NH4+-containing simulated solution and demonstrated the higher corrosion rate of pure Mg in this solution as compared to that in the pure NaCl. Cao et al. [10] confirmed that NH4+ could dissolve larger amount of MgO and Mg(OH)2 than the Cl-. All of the above studies suggest that NH4+ exhibits different effects on corrosion of magnesium from Cl- and the intrinsic mechanism of NH4+ should be clarified. ...
1
2015
... Magnesium (Mg) alloys including AZ and AM series are widely used in automotive, aircraft, electronic equipment, and other lightweight fields [[6], [7], [8], [9]]. However, the poor corrosion resistance is the most important property that limits the application of these alloys. Previous investigations have paid much attention on the corrosion of magnesium alloys in the presence of Cl- in atmospheric [[10], [11], [12], [13], [14]] and aqueous environments [[15], [16], [17], [18], [19], [20]]. However, the influence of NH4+ on the corrosion of magnesium alloys is underappreciated. Volovitch et al. [21] compared the corrosion mechanisms of Zn (Mg, Al) coated steel in accelerated tests and field exposure and suggested that the solution containing NH4+ exhibited a preponderant correlation between the laboratory simulation and natural exposure test. Battocchi et al. [22] also built the NH4+-containing simulated solution and demonstrated the higher corrosion rate of pure Mg in this solution as compared to that in the pure NaCl. Cao et al. [10] confirmed that NH4+ could dissolve larger amount of MgO and Mg(OH)2 than the Cl-. All of the above studies suggest that NH4+ exhibits different effects on corrosion of magnesium from Cl- and the intrinsic mechanism of NH4+ should be clarified. ...
4
2016
... Magnesium (Mg) alloys including AZ and AM series are widely used in automotive, aircraft, electronic equipment, and other lightweight fields [[6], [7], [8], [9]]. However, the poor corrosion resistance is the most important property that limits the application of these alloys. Previous investigations have paid much attention on the corrosion of magnesium alloys in the presence of Cl- in atmospheric [[10], [11], [12], [13], [14]] and aqueous environments [[15], [16], [17], [18], [19], [20]]. However, the influence of NH4+ on the corrosion of magnesium alloys is underappreciated. Volovitch et al. [21] compared the corrosion mechanisms of Zn (Mg, Al) coated steel in accelerated tests and field exposure and suggested that the solution containing NH4+ exhibited a preponderant correlation between the laboratory simulation and natural exposure test. Battocchi et al. [22] also built the NH4+-containing simulated solution and demonstrated the higher corrosion rate of pure Mg in this solution as compared to that in the pure NaCl. Cao et al. [10] confirmed that NH4+ could dissolve larger amount of MgO and Mg(OH)2 than the Cl-. All of the above studies suggest that NH4+ exhibits different effects on corrosion of magnesium from Cl- and the intrinsic mechanism of NH4+ should be clarified. ...
... Fig. 4 shows the FTIR (a) and XRD (b) spectra of the corrosion products formed on AZ31 magnesium alloy after immersion in different solutions for 120 h. The broad bands around 1640 cm-1 and 3436 cm-1 are attributed to the bending vibrations and stretching vibrations of H2O and H2O/OH- groups, respectively [34]. The peaks around 1400 cm-1 is ascribed to the presence of CO32- [35]. The peak at 1100 cm-1 and the associated peaks at lower wavenumbers can be assigned to the presence of Mg5(CO3)4(OH)2·xH2O (x = 4 or 5) [36]. The peaks in the XRD spectra are not obvious because the surface corrosion products are easy to be peeled off. Even so, the peaks centered at 11.1°, 22.2°, and 60.8° imply the existence of aluminum magnesium hydroxide carbonate hydrate ((Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O) [13]. The corrosion product powders dropped from the specimen are also collected and examined by XRD (Fig. S2, in Supporting Information), and the extra peaks indicate the presence of Mg5(CO3)4(OH)2·5H2O. ...
... XRD analysis implies the presence of Mg-Al-CO32- hydrotalcite-like compound (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O (HTLc) and Mg5(CO3)4(OH)2·5H2O (Section 3.2). The presence of Mg5(CO3)4(OH)2·5H2O is likely attributed to the diffusion of CO2 from the environment, which reacts with unstable Mg(OH)2 [52,53]. Arrabal et al. [54,55] found the presence of (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O on the magnesium alloy after immersion in 3.5 wt.% NaCl. Jang et al. [56] also found that the (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O was the primary corrosion product on AZ31 alloy in simulated physiological solutions. In atmospheric environment, Liao et al. [13] demonstrated the existence of the HTLc as the dominant corrosion products and proposed a simplified formation process. They considered that the high temperature on sunny days and the high local pH promoted the dissolution of Al3+ ions and thus the (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O was developed. In the present work, the solution is acidic in the initial immersion period in the presence of NH4+. This could accelerate the dissolution of Al to Al3+ and enhance the formation of (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O in the later immersion stage, during which the formation of Al-rich precursors of hydrotalcite (HT) and exchange of OH- for CO32- are feasible [57]. This HT-like compound is similar to HT and presents a structure related to brucite with the general formula [Mg1-x2+Mx3+(OH)2]x+Ax/nn-·mH2O, where M2+ and M3+ are divalent and trivalent metals, respectively, and An- is an anion [13,57]. ...
... is an anion [13,57]. ...
1
2019
... Magnesium (Mg) alloys including AZ and AM series are widely used in automotive, aircraft, electronic equipment, and other lightweight fields [[6], [7], [8], [9]]. However, the poor corrosion resistance is the most important property that limits the application of these alloys. Previous investigations have paid much attention on the corrosion of magnesium alloys in the presence of Cl- in atmospheric [[10], [11], [12], [13], [14]] and aqueous environments [[15], [16], [17], [18], [19], [20]]. However, the influence of NH4+ on the corrosion of magnesium alloys is underappreciated. Volovitch et al. [21] compared the corrosion mechanisms of Zn (Mg, Al) coated steel in accelerated tests and field exposure and suggested that the solution containing NH4+ exhibited a preponderant correlation between the laboratory simulation and natural exposure test. Battocchi et al. [22] also built the NH4+-containing simulated solution and demonstrated the higher corrosion rate of pure Mg in this solution as compared to that in the pure NaCl. Cao et al. [10] confirmed that NH4+ could dissolve larger amount of MgO and Mg(OH)2 than the Cl-. All of the above studies suggest that NH4+ exhibits different effects on corrosion of magnesium from Cl- and the intrinsic mechanism of NH4+ should be clarified. ...
1
2002
... Magnesium (Mg) alloys including AZ and AM series are widely used in automotive, aircraft, electronic equipment, and other lightweight fields [[6], [7], [8], [9]]. However, the poor corrosion resistance is the most important property that limits the application of these alloys. Previous investigations have paid much attention on the corrosion of magnesium alloys in the presence of Cl- in atmospheric [[10], [11], [12], [13], [14]] and aqueous environments [[15], [16], [17], [18], [19], [20]]. However, the influence of NH4+ on the corrosion of magnesium alloys is underappreciated. Volovitch et al. [21] compared the corrosion mechanisms of Zn (Mg, Al) coated steel in accelerated tests and field exposure and suggested that the solution containing NH4+ exhibited a preponderant correlation between the laboratory simulation and natural exposure test. Battocchi et al. [22] also built the NH4+-containing simulated solution and demonstrated the higher corrosion rate of pure Mg in this solution as compared to that in the pure NaCl. Cao et al. [10] confirmed that NH4+ could dissolve larger amount of MgO and Mg(OH)2 than the Cl-. All of the above studies suggest that NH4+ exhibits different effects on corrosion of magnesium from Cl- and the intrinsic mechanism of NH4+ should be clarified. ...
2
2008
... Magnesium (Mg) alloys including AZ and AM series are widely used in automotive, aircraft, electronic equipment, and other lightweight fields [[6], [7], [8], [9]]. However, the poor corrosion resistance is the most important property that limits the application of these alloys. Previous investigations have paid much attention on the corrosion of magnesium alloys in the presence of Cl- in atmospheric [[10], [11], [12], [13], [14]] and aqueous environments [[15], [16], [17], [18], [19], [20]]. However, the influence of NH4+ on the corrosion of magnesium alloys is underappreciated. Volovitch et al. [21] compared the corrosion mechanisms of Zn (Mg, Al) coated steel in accelerated tests and field exposure and suggested that the solution containing NH4+ exhibited a preponderant correlation between the laboratory simulation and natural exposure test. Battocchi et al. [22] also built the NH4+-containing simulated solution and demonstrated the higher corrosion rate of pure Mg in this solution as compared to that in the pure NaCl. Cao et al. [10] confirmed that NH4+ could dissolve larger amount of MgO and Mg(OH)2 than the Cl-. All of the above studies suggest that NH4+ exhibits different effects on corrosion of magnesium from Cl- and the intrinsic mechanism of NH4+ should be clarified. ...
... was hydrogen evolution volume (mL), t was the immersion time (day), n was the period of immersion. The average corrosion rate, PH (mm a-1), was evaluated through the following equation [16,33]: ...
1
1997
... Magnesium (Mg) alloys including AZ and AM series are widely used in automotive, aircraft, electronic equipment, and other lightweight fields [[6], [7], [8], [9]]. However, the poor corrosion resistance is the most important property that limits the application of these alloys. Previous investigations have paid much attention on the corrosion of magnesium alloys in the presence of Cl- in atmospheric [[10], [11], [12], [13], [14]] and aqueous environments [[15], [16], [17], [18], [19], [20]]. However, the influence of NH4+ on the corrosion of magnesium alloys is underappreciated. Volovitch et al. [21] compared the corrosion mechanisms of Zn (Mg, Al) coated steel in accelerated tests and field exposure and suggested that the solution containing NH4+ exhibited a preponderant correlation between the laboratory simulation and natural exposure test. Battocchi et al. [22] also built the NH4+-containing simulated solution and demonstrated the higher corrosion rate of pure Mg in this solution as compared to that in the pure NaCl. Cao et al. [10] confirmed that NH4+ could dissolve larger amount of MgO and Mg(OH)2 than the Cl-. All of the above studies suggest that NH4+ exhibits different effects on corrosion of magnesium from Cl- and the intrinsic mechanism of NH4+ should be clarified. ...
1
2008
... Magnesium (Mg) alloys including AZ and AM series are widely used in automotive, aircraft, electronic equipment, and other lightweight fields [[6], [7], [8], [9]]. However, the poor corrosion resistance is the most important property that limits the application of these alloys. Previous investigations have paid much attention on the corrosion of magnesium alloys in the presence of Cl- in atmospheric [[10], [11], [12], [13], [14]] and aqueous environments [[15], [16], [17], [18], [19], [20]]. However, the influence of NH4+ on the corrosion of magnesium alloys is underappreciated. Volovitch et al. [21] compared the corrosion mechanisms of Zn (Mg, Al) coated steel in accelerated tests and field exposure and suggested that the solution containing NH4+ exhibited a preponderant correlation between the laboratory simulation and natural exposure test. Battocchi et al. [22] also built the NH4+-containing simulated solution and demonstrated the higher corrosion rate of pure Mg in this solution as compared to that in the pure NaCl. Cao et al. [10] confirmed that NH4+ could dissolve larger amount of MgO and Mg(OH)2 than the Cl-. All of the above studies suggest that NH4+ exhibits different effects on corrosion of magnesium from Cl- and the intrinsic mechanism of NH4+ should be clarified. ...
1
2013
... Magnesium (Mg) alloys including AZ and AM series are widely used in automotive, aircraft, electronic equipment, and other lightweight fields [[6], [7], [8], [9]]. However, the poor corrosion resistance is the most important property that limits the application of these alloys. Previous investigations have paid much attention on the corrosion of magnesium alloys in the presence of Cl- in atmospheric [[10], [11], [12], [13], [14]] and aqueous environments [[15], [16], [17], [18], [19], [20]]. However, the influence of NH4+ on the corrosion of magnesium alloys is underappreciated. Volovitch et al. [21] compared the corrosion mechanisms of Zn (Mg, Al) coated steel in accelerated tests and field exposure and suggested that the solution containing NH4+ exhibited a preponderant correlation between the laboratory simulation and natural exposure test. Battocchi et al. [22] also built the NH4+-containing simulated solution and demonstrated the higher corrosion rate of pure Mg in this solution as compared to that in the pure NaCl. Cao et al. [10] confirmed that NH4+ could dissolve larger amount of MgO and Mg(OH)2 than the Cl-. All of the above studies suggest that NH4+ exhibits different effects on corrosion of magnesium from Cl- and the intrinsic mechanism of NH4+ should be clarified. ...
1
2018
... Magnesium (Mg) alloys including AZ and AM series are widely used in automotive, aircraft, electronic equipment, and other lightweight fields [[6], [7], [8], [9]]. However, the poor corrosion resistance is the most important property that limits the application of these alloys. Previous investigations have paid much attention on the corrosion of magnesium alloys in the presence of Cl- in atmospheric [[10], [11], [12], [13], [14]] and aqueous environments [[15], [16], [17], [18], [19], [20]]. However, the influence of NH4+ on the corrosion of magnesium alloys is underappreciated. Volovitch et al. [21] compared the corrosion mechanisms of Zn (Mg, Al) coated steel in accelerated tests and field exposure and suggested that the solution containing NH4+ exhibited a preponderant correlation between the laboratory simulation and natural exposure test. Battocchi et al. [22] also built the NH4+-containing simulated solution and demonstrated the higher corrosion rate of pure Mg in this solution as compared to that in the pure NaCl. Cao et al. [10] confirmed that NH4+ could dissolve larger amount of MgO and Mg(OH)2 than the Cl-. All of the above studies suggest that NH4+ exhibits different effects on corrosion of magnesium from Cl- and the intrinsic mechanism of NH4+ should be clarified. ...
1
2015
... Magnesium (Mg) alloys including AZ and AM series are widely used in automotive, aircraft, electronic equipment, and other lightweight fields [[6], [7], [8], [9]]. However, the poor corrosion resistance is the most important property that limits the application of these alloys. Previous investigations have paid much attention on the corrosion of magnesium alloys in the presence of Cl- in atmospheric [[10], [11], [12], [13], [14]] and aqueous environments [[15], [16], [17], [18], [19], [20]]. However, the influence of NH4+ on the corrosion of magnesium alloys is underappreciated. Volovitch et al. [21] compared the corrosion mechanisms of Zn (Mg, Al) coated steel in accelerated tests and field exposure and suggested that the solution containing NH4+ exhibited a preponderant correlation between the laboratory simulation and natural exposure test. Battocchi et al. [22] also built the NH4+-containing simulated solution and demonstrated the higher corrosion rate of pure Mg in this solution as compared to that in the pure NaCl. Cao et al. [10] confirmed that NH4+ could dissolve larger amount of MgO and Mg(OH)2 than the Cl-. All of the above studies suggest that NH4+ exhibits different effects on corrosion of magnesium from Cl- and the intrinsic mechanism of NH4+ should be clarified. ...
1
2006
... Magnesium (Mg) alloys including AZ and AM series are widely used in automotive, aircraft, electronic equipment, and other lightweight fields [[6], [7], [8], [9]]. However, the poor corrosion resistance is the most important property that limits the application of these alloys. Previous investigations have paid much attention on the corrosion of magnesium alloys in the presence of Cl- in atmospheric [[10], [11], [12], [13], [14]] and aqueous environments [[15], [16], [17], [18], [19], [20]]. However, the influence of NH4+ on the corrosion of magnesium alloys is underappreciated. Volovitch et al. [21] compared the corrosion mechanisms of Zn (Mg, Al) coated steel in accelerated tests and field exposure and suggested that the solution containing NH4+ exhibited a preponderant correlation between the laboratory simulation and natural exposure test. Battocchi et al. [22] also built the NH4+-containing simulated solution and demonstrated the higher corrosion rate of pure Mg in this solution as compared to that in the pure NaCl. Cao et al. [10] confirmed that NH4+ could dissolve larger amount of MgO and Mg(OH)2 than the Cl-. All of the above studies suggest that NH4+ exhibits different effects on corrosion of magnesium from Cl- and the intrinsic mechanism of NH4+ should be clarified. ...
1
2015
... The effect of NH4+ on the corrosion of metallic materials can be summarized as the following three aspects: complexation with metallic ions, modification of solution, and disruption of corrosion products. Firstly, it has been found that the corrosion of Cu and Zn was accelerated in the presence of NH4+, attributed to the formation of soluble complex compounds [[23], [24], [25]]. Secondly, the solution chemistry is modified in the presence of NH4+, of which the concentration is pH-dependent [26]. Moreover, the buffering effect of NH4+ can provide H+ and support the cathodic reactions. Cui et al. [27] found that the buffering effect of NH4+ combined with the effect of NO3- and Cl- resulted in the “autocatalytic” pitting corrosion of AZ31 magnesium alloy. Thirdly, the corrosion products can be significantly deteriorated by NH4+. Buggio et al. [28] reported that metastability of hydride phases was favored in the presence of NH4+ which could alter the equilibrium dissociation of water. Citterio et al. [29] declared that the dissociation of NH4+ into NH3 and H+ hindered the precipitation of Mg(OH)2. Cao et al. [30] proposed that NH4+ could dissolve the relatively protective MgO layer and provide a path for the aggressive electrolytes to diffuse to the substrate. ...
1
1993
... The effect of NH4+ on the corrosion of metallic materials can be summarized as the following three aspects: complexation with metallic ions, modification of solution, and disruption of corrosion products. Firstly, it has been found that the corrosion of Cu and Zn was accelerated in the presence of NH4+, attributed to the formation of soluble complex compounds [[23], [24], [25]]. Secondly, the solution chemistry is modified in the presence of NH4+, of which the concentration is pH-dependent [26]. Moreover, the buffering effect of NH4+ can provide H+ and support the cathodic reactions. Cui et al. [27] found that the buffering effect of NH4+ combined with the effect of NO3- and Cl- resulted in the “autocatalytic” pitting corrosion of AZ31 magnesium alloy. Thirdly, the corrosion products can be significantly deteriorated by NH4+. Buggio et al. [28] reported that metastability of hydride phases was favored in the presence of NH4+ which could alter the equilibrium dissociation of water. Citterio et al. [29] declared that the dissociation of NH4+ into NH3 and H+ hindered the precipitation of Mg(OH)2. Cao et al. [30] proposed that NH4+ could dissolve the relatively protective MgO layer and provide a path for the aggressive electrolytes to diffuse to the substrate. ...
1
2005
... The effect of NH4+ on the corrosion of metallic materials can be summarized as the following three aspects: complexation with metallic ions, modification of solution, and disruption of corrosion products. Firstly, it has been found that the corrosion of Cu and Zn was accelerated in the presence of NH4+, attributed to the formation of soluble complex compounds [[23], [24], [25]]. Secondly, the solution chemistry is modified in the presence of NH4+, of which the concentration is pH-dependent [26]. Moreover, the buffering effect of NH4+ can provide H+ and support the cathodic reactions. Cui et al. [27] found that the buffering effect of NH4+ combined with the effect of NO3- and Cl- resulted in the “autocatalytic” pitting corrosion of AZ31 magnesium alloy. Thirdly, the corrosion products can be significantly deteriorated by NH4+. Buggio et al. [28] reported that metastability of hydride phases was favored in the presence of NH4+ which could alter the equilibrium dissociation of water. Citterio et al. [29] declared that the dissociation of NH4+ into NH3 and H+ hindered the precipitation of Mg(OH)2. Cao et al. [30] proposed that NH4+ could dissolve the relatively protective MgO layer and provide a path for the aggressive electrolytes to diffuse to the substrate. ...
1
2001
... The effect of NH4+ on the corrosion of metallic materials can be summarized as the following three aspects: complexation with metallic ions, modification of solution, and disruption of corrosion products. Firstly, it has been found that the corrosion of Cu and Zn was accelerated in the presence of NH4+, attributed to the formation of soluble complex compounds [[23], [24], [25]]. Secondly, the solution chemistry is modified in the presence of NH4+, of which the concentration is pH-dependent [26]. Moreover, the buffering effect of NH4+ can provide H+ and support the cathodic reactions. Cui et al. [27] found that the buffering effect of NH4+ combined with the effect of NO3- and Cl- resulted in the “autocatalytic” pitting corrosion of AZ31 magnesium alloy. Thirdly, the corrosion products can be significantly deteriorated by NH4+. Buggio et al. [28] reported that metastability of hydride phases was favored in the presence of NH4+ which could alter the equilibrium dissociation of water. Citterio et al. [29] declared that the dissociation of NH4+ into NH3 and H+ hindered the precipitation of Mg(OH)2. Cao et al. [30] proposed that NH4+ could dissolve the relatively protective MgO layer and provide a path for the aggressive electrolytes to diffuse to the substrate. ...
4
2018
... The effect of NH4+ on the corrosion of metallic materials can be summarized as the following three aspects: complexation with metallic ions, modification of solution, and disruption of corrosion products. Firstly, it has been found that the corrosion of Cu and Zn was accelerated in the presence of NH4+, attributed to the formation of soluble complex compounds [[23], [24], [25]]. Secondly, the solution chemistry is modified in the presence of NH4+, of which the concentration is pH-dependent [26]. Moreover, the buffering effect of NH4+ can provide H+ and support the cathodic reactions. Cui et al. [27] found that the buffering effect of NH4+ combined with the effect of NO3- and Cl- resulted in the “autocatalytic” pitting corrosion of AZ31 magnesium alloy. Thirdly, the corrosion products can be significantly deteriorated by NH4+. Buggio et al. [28] reported that metastability of hydride phases was favored in the presence of NH4+ which could alter the equilibrium dissociation of water. Citterio et al. [29] declared that the dissociation of NH4+ into NH3 and H+ hindered the precipitation of Mg(OH)2. Cao et al. [30] proposed that NH4+ could dissolve the relatively protective MgO layer and provide a path for the aggressive electrolytes to diffuse to the substrate. ...
... Fig. 11 compares the corrosion rate obtained by weight loss, hydrogen evolution, and EIS after immersion for different time. A general decrease of corrosion rate in the three solutions is observed despite some fluctuations in dilute environments. The decrease of corrosion rate with increasing immersion can be ascribed to the increase of solution pH [41,42], formation of corrosion products which decreases the active surface area [[43], [44], [45]], and the decrease of NH4+ content at high pH environment [27]. The fluctuation of corrosion rate in solution with 0.001 M NH4+ is attributed to the local rupture of the surface film [10,41]. ...
... Cui et al. [27] reported that the amount of NH4+ is more than 90% in a solution with a pH less than 8. Therefore, NH4+ can influence the whole corrosion process of AZ31 magnesium alloy. Firstly, the complexation of NH4+ is excluded because the complexing action between NH4+ and Mg is negligible and the Zn in AZ31 alloy is limited to cause a considerable complexation [27,48]. Even so, the presence of NH4+ may affect the surface corrosion products and expedite the corrosion processes. In Fig. 1(c), the pH values of all solutions after a 5-day immersion are below 8.4, under which the stable Mg(OH)2 layer is difficult to be preserved because the precipitation of brucite is possible when the pH exceeds 9.43 [49]. This has been confirmed by XRD and FTIR results (Fig. 4) where the peak from Mg(OH)2 is not detected. Additionally, Cao et al. [30] reported that the acceleration effect of NH4+ on Mg corrosion could be attributed to the dissolution of the thin protective inner MgO layer/outer non-protective Mg(OH)2 layer in the corrosion product film. The dissolution reactions are as follows: ...
... and Mg is negligible and the Zn in AZ31 alloy is limited to cause a considerable complexation [27,48]. Even so, the presence of NH4+ may affect the surface corrosion products and expedite the corrosion processes. In Fig. 1(c), the pH values of all solutions after a 5-day immersion are below 8.4, under which the stable Mg(OH)2 layer is difficult to be preserved because the precipitation of brucite is possible when the pH exceeds 9.43 [49]. This has been confirmed by XRD and FTIR results (Fig. 4) where the peak from Mg(OH)2 is not detected. Additionally, Cao et al. [30] reported that the acceleration effect of NH4+ on Mg corrosion could be attributed to the dissolution of the thin protective inner MgO layer/outer non-protective Mg(OH)2 layer in the corrosion product film. The dissolution reactions are as follows: ...
4
2016
... The effect of NH4+ on the corrosion of metallic materials can be summarized as the following three aspects: complexation with metallic ions, modification of solution, and disruption of corrosion products. Firstly, it has been found that the corrosion of Cu and Zn was accelerated in the presence of NH4+, attributed to the formation of soluble complex compounds [[23], [24], [25]]. Secondly, the solution chemistry is modified in the presence of NH4+, of which the concentration is pH-dependent [26]. Moreover, the buffering effect of NH4+ can provide H+ and support the cathodic reactions. Cui et al. [27] found that the buffering effect of NH4+ combined with the effect of NO3- and Cl- resulted in the “autocatalytic” pitting corrosion of AZ31 magnesium alloy. Thirdly, the corrosion products can be significantly deteriorated by NH4+. Buggio et al. [28] reported that metastability of hydride phases was favored in the presence of NH4+ which could alter the equilibrium dissociation of water. Citterio et al. [29] declared that the dissociation of NH4+ into NH3 and H+ hindered the precipitation of Mg(OH)2. Cao et al. [30] proposed that NH4+ could dissolve the relatively protective MgO layer and provide a path for the aggressive electrolytes to diffuse to the substrate. ...
... Notably, there exist dissimilarities between the corrosion rates obtained by different methods because of the intrinsic differences of these approaches. Generally, the corrosion rate calculated by hydrogen evolution is relatively lower as compared to other techniques. On one hand, the increase in the released hydrogen volume is low in the final stage and is difficult to differentiate. On the other hand, formation of hydride sub-products consumes the generated H2 and decreases the apparent corrosion rate [5,28,46]. Buggio et al. [28] revealed that the chemical recombination of hydrogen atoms of oxidic metastable hydrides was the kinetically determinant process during anodic oxidation of Mg in NH4+-containing electrolytes. In the 0.001 M NH4+ solution, corrosion rate derived from weight loss method is the highest, which may be attributed to the Non-Faraday process, including the undermining of second phases [47] and the dissolution of univalent magnesium ions [33], that cannot be detected by electrochemical methods. In 0.01 M and 0.1 M NH4+ solution, corrosion rate obtained by EIS is higher than that obtained by weight loss, especially in the 0.1 M NH4+ solution. Several reasons may be responsible for this phenomenon. Firstly, the electrochemical dissolution process may overshadow the Non-Faraday process, yielding a higher corrosion rate obtained by EIS. Secondly, EIS is an instantaneous approach, which provides a corrosion rate at a particular time over which the test is performed [8]. Therefore, this rate may be higher than the average corrosion rate that extracted from mass-loss method. ...
... ]. Buggio et al. [28] revealed that the chemical recombination of hydrogen atoms of oxidic metastable hydrides was the kinetically determinant process during anodic oxidation of Mg in NH4+-containing electrolytes. In the 0.001 M NH4+ solution, corrosion rate derived from weight loss method is the highest, which may be attributed to the Non-Faraday process, including the undermining of second phases [47] and the dissolution of univalent magnesium ions [33], that cannot be detected by electrochemical methods. In 0.01 M and 0.1 M NH4+ solution, corrosion rate obtained by EIS is higher than that obtained by weight loss, especially in the 0.1 M NH4+ solution. Several reasons may be responsible for this phenomenon. Firstly, the electrochemical dissolution process may overshadow the Non-Faraday process, yielding a higher corrosion rate obtained by EIS. Secondly, EIS is an instantaneous approach, which provides a corrosion rate at a particular time over which the test is performed [8]. Therefore, this rate may be higher than the average corrosion rate that extracted from mass-loss method. ...
... As reported by Buggio et al. [28], the microstructure of Mg alloy, the nature of the second salt or its proportion appeared to be unimportant as the anodic (or cathodic) potential was raised in NH4+-based solution. Pébère et al. [45] investigated the electrochemical behavior of magnesium using SO42- as a supporting electrolyte because of the low aggressive of SO42-. Cui et al. [58] reported that SO42- ions had no obvious effect on the icorr in saline solution. Zeng et al. [42] reported that SO42- exhibited little effect on the corrosion of magnesium alloy. Therefore, compared with the strong effect of NH4+ ion, the effect of SO42- on Mg corrosion is negligible. Potentiodynamic polarization curves of AZ31 magnesium in 0.005 M (NH4)2SO4 and Na2SO4 (pH values adjusted with H2SO4) are shown in Fig. 13 and the extracted parameters are listed in Table 4. As shown in Fig. 13, the cathodic Tafel slope of the polarization curve increases with the addition of NH4+. Moreover, there is a pseudo-passive region at the anodic branch of AZ31 in all Na2SO4 solutions. Therefore, Fig. 13 indicates that the protective MgO film of the surface could be dissolved by the NH4+. However, the influence of H+ on the corrosion behavior of AZ31 magnesium can be neglected. In view of this concentration, the effect of NH4+ on the corrosion rate of the AZ31 magnesium alloy is over 88%. Similar results are also found when comparing the polarization curves in 0.05 M (NH4)2SO4 and Na2SO4 with the same original pH (Fig. S3, in Supporting Information). ...
1
2014
... The effect of NH4+ on the corrosion of metallic materials can be summarized as the following three aspects: complexation with metallic ions, modification of solution, and disruption of corrosion products. Firstly, it has been found that the corrosion of Cu and Zn was accelerated in the presence of NH4+, attributed to the formation of soluble complex compounds [[23], [24], [25]]. Secondly, the solution chemistry is modified in the presence of NH4+, of which the concentration is pH-dependent [26]. Moreover, the buffering effect of NH4+ can provide H+ and support the cathodic reactions. Cui et al. [27] found that the buffering effect of NH4+ combined with the effect of NO3- and Cl- resulted in the “autocatalytic” pitting corrosion of AZ31 magnesium alloy. Thirdly, the corrosion products can be significantly deteriorated by NH4+. Buggio et al. [28] reported that metastability of hydride phases was favored in the presence of NH4+ which could alter the equilibrium dissociation of water. Citterio et al. [29] declared that the dissociation of NH4+ into NH3 and H+ hindered the precipitation of Mg(OH)2. Cao et al. [30] proposed that NH4+ could dissolve the relatively protective MgO layer and provide a path for the aggressive electrolytes to diffuse to the substrate. ...
4
2019
... The effect of NH4+ on the corrosion of metallic materials can be summarized as the following three aspects: complexation with metallic ions, modification of solution, and disruption of corrosion products. Firstly, it has been found that the corrosion of Cu and Zn was accelerated in the presence of NH4+, attributed to the formation of soluble complex compounds [[23], [24], [25]]. Secondly, the solution chemistry is modified in the presence of NH4+, of which the concentration is pH-dependent [26]. Moreover, the buffering effect of NH4+ can provide H+ and support the cathodic reactions. Cui et al. [27] found that the buffering effect of NH4+ combined with the effect of NO3- and Cl- resulted in the “autocatalytic” pitting corrosion of AZ31 magnesium alloy. Thirdly, the corrosion products can be significantly deteriorated by NH4+. Buggio et al. [28] reported that metastability of hydride phases was favored in the presence of NH4+ which could alter the equilibrium dissociation of water. Citterio et al. [29] declared that the dissociation of NH4+ into NH3 and H+ hindered the precipitation of Mg(OH)2. Cao et al. [30] proposed that NH4+ could dissolve the relatively protective MgO layer and provide a path for the aggressive electrolytes to diffuse to the substrate. ...
... Cui et al. [27] reported that the amount of NH4+ is more than 90% in a solution with a pH less than 8. Therefore, NH4+ can influence the whole corrosion process of AZ31 magnesium alloy. Firstly, the complexation of NH4+ is excluded because the complexing action between NH4+ and Mg is negligible and the Zn in AZ31 alloy is limited to cause a considerable complexation [27,48]. Even so, the presence of NH4+ may affect the surface corrosion products and expedite the corrosion processes. In Fig. 1(c), the pH values of all solutions after a 5-day immersion are below 8.4, under which the stable Mg(OH)2 layer is difficult to be preserved because the precipitation of brucite is possible when the pH exceeds 9.43 [49]. This has been confirmed by XRD and FTIR results (Fig. 4) where the peak from Mg(OH)2 is not detected. Additionally, Cao et al. [30] reported that the acceleration effect of NH4+ on Mg corrosion could be attributed to the dissolution of the thin protective inner MgO layer/outer non-protective Mg(OH)2 layer in the corrosion product film. The dissolution reactions are as follows: ...
... Moreover, ammonium ion preferentially degrades MgO, which is assumed to be more protective than Mg(OH)2 [30,43,50]. Actually, there must be a thin Mg(OH)2 and MgO film formed at the interface of solution and magnesium, which may be caused by the partial alkalization on account of the cathodic reactions during dissolution of magnesium [51]. Due to the dissolution of the corrosion product layer, a fresh surface of AZ31 magnesium alloy is exposed, and the substrate can directly react with the solution without any barrier. Eventually, the active surface area of AZ31 magnesium alloy decreases and the bare surface is covered by a loose corrosion product film, which may be constituted by various ions through a weak-interaction. ...
... Firstly, the two capacitive loops at high frequency and low frequency in all the three solutions can be attributed to the charge transfer process and the corrosion product film, respectively. Both of the charge transfer resistance and film resistance significantly decrease with increasing NH4+ concentration, suggesting the acceleration of the corrosion process. According to Cao et al. [30], the inner MgO layer was easily to be dissolved by NH4+ as compared to the Mg(OH)2. This creates more active surface area and results in the expedited corrosion process in the high NH4+ solutions [43,44]. With increasing immersion time, the Rct and Rf increase gradually in the 0.01 M and 0.1 M NH4+ solutions, revealing the blocking effect of the corrosion products that precipitated on the sample surface. The fluctuations of Rct and Rf in 0.001 M NH4+ solution can be ascribed to the competition between film growth and dissolution processes [41,46,60,61] on account of the slight corrosion in this environment. In addition, the variation of the capacitance values may be attributed to the variation of the film thickness and the available area. ...
2
2011
... The original weight (w0) of the samples and pH of the solutions were measured before the immersion tests. The ratio of the solution volume to the sample surface area was 30 mL/cm2. Different immersion intervals (6 h, 24 h, 48 h, 72 h, 96 h, and 120 h) were designed to obtain the corrosion kinetics in different environments. After immersion test, the corroded samples were cleaned with chromate solution (200 g L-1 CrO3 + 10 g L-1 AgNO3 + 20 g L-1 BaNO3) to remove corrosion products. Finally, the corroded samples were dried in air and reweighed, documented as wn. Five parallel experiments were taken for each group at least for obtaining accurate experimental data to check the reproducibility. The corrosion rate, VW (mg cm-2 d-1), was calculated through the following equation [31]: ...
... Fig. 10(b) shows the variations of Rp during the immersion test. A rough increase with some fluctuations is observed in 0.001 M and 0.01 M NH4+ solutions, while Rp in 0.1 M NH4+ solutions shows a monotonous increase with extending immersion time. The Rp can be transferred to corrosion current density using the Stern equation with the constant B of 60 mV [38]. Then the corrosion rate Peis (mm a-1) can be calculated through the following equation and will be discussed in the following section [31,39,40]: ...
1
2008
... was the surface area of the samples, and tn (day) was the immersion time. The average corrosion rate PW (mm a-1) was calculated by Eq. (2) [32]: ...
2
2003
... was hydrogen evolution volume (mL), t was the immersion time (day), n was the period of immersion. The average corrosion rate, PH (mm a-1), was evaluated through the following equation [16,33]: ...
... Notably, there exist dissimilarities between the corrosion rates obtained by different methods because of the intrinsic differences of these approaches. Generally, the corrosion rate calculated by hydrogen evolution is relatively lower as compared to other techniques. On one hand, the increase in the released hydrogen volume is low in the final stage and is difficult to differentiate. On the other hand, formation of hydride sub-products consumes the generated H2 and decreases the apparent corrosion rate [5,28,46]. Buggio et al. [28] revealed that the chemical recombination of hydrogen atoms of oxidic metastable hydrides was the kinetically determinant process during anodic oxidation of Mg in NH4+-containing electrolytes. In the 0.001 M NH4+ solution, corrosion rate derived from weight loss method is the highest, which may be attributed to the Non-Faraday process, including the undermining of second phases [47] and the dissolution of univalent magnesium ions [33], that cannot be detected by electrochemical methods. In 0.01 M and 0.1 M NH4+ solution, corrosion rate obtained by EIS is higher than that obtained by weight loss, especially in the 0.1 M NH4+ solution. Several reasons may be responsible for this phenomenon. Firstly, the electrochemical dissolution process may overshadow the Non-Faraday process, yielding a higher corrosion rate obtained by EIS. Secondly, EIS is an instantaneous approach, which provides a corrosion rate at a particular time over which the test is performed [8]. Therefore, this rate may be higher than the average corrosion rate that extracted from mass-loss method. ...
1
2005
... Fig. 4 shows the FTIR (a) and XRD (b) spectra of the corrosion products formed on AZ31 magnesium alloy after immersion in different solutions for 120 h. The broad bands around 1640 cm-1 and 3436 cm-1 are attributed to the bending vibrations and stretching vibrations of H2O and H2O/OH- groups, respectively [34]. The peaks around 1400 cm-1 is ascribed to the presence of CO32- [35]. The peak at 1100 cm-1 and the associated peaks at lower wavenumbers can be assigned to the presence of Mg5(CO3)4(OH)2·xH2O (x = 4 or 5) [36]. The peaks in the XRD spectra are not obvious because the surface corrosion products are easy to be peeled off. Even so, the peaks centered at 11.1°, 22.2°, and 60.8° imply the existence of aluminum magnesium hydroxide carbonate hydrate ((Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O) [13]. The corrosion product powders dropped from the specimen are also collected and examined by XRD (Fig. S2, in Supporting Information), and the extra peaks indicate the presence of Mg5(CO3)4(OH)2·5H2O. ...
1
2007
... Fig. 4 shows the FTIR (a) and XRD (b) spectra of the corrosion products formed on AZ31 magnesium alloy after immersion in different solutions for 120 h. The broad bands around 1640 cm-1 and 3436 cm-1 are attributed to the bending vibrations and stretching vibrations of H2O and H2O/OH- groups, respectively [34]. The peaks around 1400 cm-1 is ascribed to the presence of CO32- [35]. The peak at 1100 cm-1 and the associated peaks at lower wavenumbers can be assigned to the presence of Mg5(CO3)4(OH)2·xH2O (x = 4 or 5) [36]. The peaks in the XRD spectra are not obvious because the surface corrosion products are easy to be peeled off. Even so, the peaks centered at 11.1°, 22.2°, and 60.8° imply the existence of aluminum magnesium hydroxide carbonate hydrate ((Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O) [13]. The corrosion product powders dropped from the specimen are also collected and examined by XRD (Fig. S2, in Supporting Information), and the extra peaks indicate the presence of Mg5(CO3)4(OH)2·5H2O. ...
1
2018
... Fig. 4 shows the FTIR (a) and XRD (b) spectra of the corrosion products formed on AZ31 magnesium alloy after immersion in different solutions for 120 h. The broad bands around 1640 cm-1 and 3436 cm-1 are attributed to the bending vibrations and stretching vibrations of H2O and H2O/OH- groups, respectively [34]. The peaks around 1400 cm-1 is ascribed to the presence of CO32- [35]. The peak at 1100 cm-1 and the associated peaks at lower wavenumbers can be assigned to the presence of Mg5(CO3)4(OH)2·xH2O (x = 4 or 5) [36]. The peaks in the XRD spectra are not obvious because the surface corrosion products are easy to be peeled off. Even so, the peaks centered at 11.1°, 22.2°, and 60.8° imply the existence of aluminum magnesium hydroxide carbonate hydrate ((Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O) [13]. The corrosion product powders dropped from the specimen are also collected and examined by XRD (Fig. S2, in Supporting Information), and the extra peaks indicate the presence of Mg5(CO3)4(OH)2·5H2O. ...
1
2014
... Fig. 8 shows the polarization curves of the AZ31 magnesium alloy in solutions with different concentrations of NH4+ before (a) and after (b) IR compensation. The data processing of IR correction is to compensate the influence of solution resistance on the potential drop because the disparities of this parameter (obtained by EIS) among the three solutions are considerable enough to affect the polarization curves [37]. The corrosion potentials (Ecorr), cathodic Tafel slope (bc), and the corrosion current density (icorr) are extrapolated from the polarization curves and listed in Table 2. It should be noted that only cathodic polarization curves are used to conduct the Tafel fitting because the anodic part does not meet the strict prerequisites of Tafel fitting. Among them, the Ecorr corresponds to short-term OCP because the curves are scanned from OCP to positive and negative directions separately. Both the anodic and cathodic reactions are expedited with increasing NH4+ concentration, yielding an overall curve shift to the lower right direction. This also leads to the negative shift of the Ecorr from -1.524 to -1.722 VSCE, which is consistent with the long-term OCP evolutions (Fig. 7(a)). Moreover, the corrosion current density increases almost 70 times as compared the values in the 0.001 M and 0.1 M NH4+ solutions (Table 2). ...
1
1990
... Fig. 10(b) shows the variations of Rp during the immersion test. A rough increase with some fluctuations is observed in 0.001 M and 0.01 M NH4+ solutions, while Rp in 0.1 M NH4+ solutions shows a monotonous increase with extending immersion time. The Rp can be transferred to corrosion current density using the Stern equation with the constant B of 60 mV [38]. Then the corrosion rate Peis (mm a-1) can be calculated through the following equation and will be discussed in the following section [31,39,40]: ...
1
1992
... Fig. 10(b) shows the variations of Rp during the immersion test. A rough increase with some fluctuations is observed in 0.001 M and 0.01 M NH4+ solutions, while Rp in 0.1 M NH4+ solutions shows a monotonous increase with extending immersion time. The Rp can be transferred to corrosion current density using the Stern equation with the constant B of 60 mV [38]. Then the corrosion rate Peis (mm a-1) can be calculated through the following equation and will be discussed in the following section [31,39,40]: ...
1
2010
... Fig. 10(b) shows the variations of Rp during the immersion test. A rough increase with some fluctuations is observed in 0.001 M and 0.01 M NH4+ solutions, while Rp in 0.1 M NH4+ solutions shows a monotonous increase with extending immersion time. The Rp can be transferred to corrosion current density using the Stern equation with the constant B of 60 mV [38]. Then the corrosion rate Peis (mm a-1) can be calculated through the following equation and will be discussed in the following section [31,39,40]: ...
3
2014
... Fig. 11 compares the corrosion rate obtained by weight loss, hydrogen evolution, and EIS after immersion for different time. A general decrease of corrosion rate in the three solutions is observed despite some fluctuations in dilute environments. The decrease of corrosion rate with increasing immersion can be ascribed to the increase of solution pH [41,42], formation of corrosion products which decreases the active surface area [[43], [44], [45]], and the decrease of NH4+ content at high pH environment [27]. The fluctuation of corrosion rate in solution with 0.001 M NH4+ is attributed to the local rupture of the surface film [10,41]. ...
... ,41]. ...
... Firstly, the two capacitive loops at high frequency and low frequency in all the three solutions can be attributed to the charge transfer process and the corrosion product film, respectively. Both of the charge transfer resistance and film resistance significantly decrease with increasing NH4+ concentration, suggesting the acceleration of the corrosion process. According to Cao et al. [30], the inner MgO layer was easily to be dissolved by NH4+ as compared to the Mg(OH)2. This creates more active surface area and results in the expedited corrosion process in the high NH4+ solutions [43,44]. With increasing immersion time, the Rct and Rf increase gradually in the 0.01 M and 0.1 M NH4+ solutions, revealing the blocking effect of the corrosion products that precipitated on the sample surface. The fluctuations of Rct and Rf in 0.001 M NH4+ solution can be ascribed to the competition between film growth and dissolution processes [41,46,60,61] on account of the slight corrosion in this environment. In addition, the variation of the capacitance values may be attributed to the variation of the film thickness and the available area. ...
2
2014
... Fig. 11 compares the corrosion rate obtained by weight loss, hydrogen evolution, and EIS after immersion for different time. A general decrease of corrosion rate in the three solutions is observed despite some fluctuations in dilute environments. The decrease of corrosion rate with increasing immersion can be ascribed to the increase of solution pH [41,42], formation of corrosion products which decreases the active surface area [[43], [44], [45]], and the decrease of NH4+ content at high pH environment [27]. The fluctuation of corrosion rate in solution with 0.001 M NH4+ is attributed to the local rupture of the surface film [10,41]. ...
... As reported by Buggio et al. [28], the microstructure of Mg alloy, the nature of the second salt or its proportion appeared to be unimportant as the anodic (or cathodic) potential was raised in NH4+-based solution. Pébère et al. [45] investigated the electrochemical behavior of magnesium using SO42- as a supporting electrolyte because of the low aggressive of SO42-. Cui et al. [58] reported that SO42- ions had no obvious effect on the icorr in saline solution. Zeng et al. [42] reported that SO42- exhibited little effect on the corrosion of magnesium alloy. Therefore, compared with the strong effect of NH4+ ion, the effect of SO42- on Mg corrosion is negligible. Potentiodynamic polarization curves of AZ31 magnesium in 0.005 M (NH4)2SO4 and Na2SO4 (pH values adjusted with H2SO4) are shown in Fig. 13 and the extracted parameters are listed in Table 4. As shown in Fig. 13, the cathodic Tafel slope of the polarization curve increases with the addition of NH4+. Moreover, there is a pseudo-passive region at the anodic branch of AZ31 in all Na2SO4 solutions. Therefore, Fig. 13 indicates that the protective MgO film of the surface could be dissolved by the NH4+. However, the influence of H+ on the corrosion behavior of AZ31 magnesium can be neglected. In view of this concentration, the effect of NH4+ on the corrosion rate of the AZ31 magnesium alloy is over 88%. Similar results are also found when comparing the polarization curves in 0.05 M (NH4)2SO4 and Na2SO4 with the same original pH (Fig. S3, in Supporting Information). ...
4
2007
... Fig. 11 compares the corrosion rate obtained by weight loss, hydrogen evolution, and EIS after immersion for different time. A general decrease of corrosion rate in the three solutions is observed despite some fluctuations in dilute environments. The decrease of corrosion rate with increasing immersion can be ascribed to the increase of solution pH [41,42], formation of corrosion products which decreases the active surface area [[43], [44], [45]], and the decrease of NH4+ content at high pH environment [27]. The fluctuation of corrosion rate in solution with 0.001 M NH4+ is attributed to the local rupture of the surface film [10,41]. ...
... Moreover, ammonium ion preferentially degrades MgO, which is assumed to be more protective than Mg(OH)2 [30,43,50]. Actually, there must be a thin Mg(OH)2 and MgO film formed at the interface of solution and magnesium, which may be caused by the partial alkalization on account of the cathodic reactions during dissolution of magnesium [51]. Due to the dissolution of the corrosion product layer, a fresh surface of AZ31 magnesium alloy is exposed, and the substrate can directly react with the solution without any barrier. Eventually, the active surface area of AZ31 magnesium alloy decreases and the bare surface is covered by a loose corrosion product film, which may be constituted by various ions through a weak-interaction. ...
... The interfacial process occurring on the magnesium/solution interface varies with NH4+ concentration as indicated by the change of EIS loops. To intuitively present the evolution of EIS data with increasing immersion time and NH4+ concentration, the normalized impedance diagrams which are proposed by Baril et al. [43] are plotted and shown in Fig. 14. The diagrams measured in 0.001 M NH4+ solution coincide well with each other, indicating the same corrosion mechanism during the immersion test [59]. In 0.01 M NH4+ solution, the inductive loops shrink with increasing immersion time, while an additional inductive loop is detected at low frequency in the 0.1 M NH4+ solution, suggesting that aging in the solution affects the interfacial processes. Moreover, the diagrams at 0.5 h in the three solutions show distinct characteristics, revealing that NH4+ concentration also influence the dissolution process occurring on the magnesium/solution interface. ...
... Firstly, the two capacitive loops at high frequency and low frequency in all the three solutions can be attributed to the charge transfer process and the corrosion product film, respectively. Both of the charge transfer resistance and film resistance significantly decrease with increasing NH4+ concentration, suggesting the acceleration of the corrosion process. According to Cao et al. [30], the inner MgO layer was easily to be dissolved by NH4+ as compared to the Mg(OH)2. This creates more active surface area and results in the expedited corrosion process in the high NH4+ solutions [43,44]. With increasing immersion time, the Rct and Rf increase gradually in the 0.01 M and 0.1 M NH4+ solutions, revealing the blocking effect of the corrosion products that precipitated on the sample surface. The fluctuations of Rct and Rf in 0.001 M NH4+ solution can be ascribed to the competition between film growth and dissolution processes [41,46,60,61] on account of the slight corrosion in this environment. In addition, the variation of the capacitance values may be attributed to the variation of the film thickness and the available area. ...
3
2001
... Fig. 11 compares the corrosion rate obtained by weight loss, hydrogen evolution, and EIS after immersion for different time. A general decrease of corrosion rate in the three solutions is observed despite some fluctuations in dilute environments. The decrease of corrosion rate with increasing immersion can be ascribed to the increase of solution pH [41,42], formation of corrosion products which decreases the active surface area [[43], [44], [45]], and the decrease of NH4+ content at high pH environment [27]. The fluctuation of corrosion rate in solution with 0.001 M NH4+ is attributed to the local rupture of the surface film [10,41]. ...
... The cathodic current associated with the reduction of oxygen can be neglected since the oxygen reduction is not important to Mg corrosion [44]. At the initial immersion stage, hydrolysis of NH4+ results in an acidic environment and promotes the cathodic process. In NH4+-containing solution, the anodic process is very complex. The reactions are as follows: ...
... Firstly, the two capacitive loops at high frequency and low frequency in all the three solutions can be attributed to the charge transfer process and the corrosion product film, respectively. Both of the charge transfer resistance and film resistance significantly decrease with increasing NH4+ concentration, suggesting the acceleration of the corrosion process. According to Cao et al. [30], the inner MgO layer was easily to be dissolved by NH4+ as compared to the Mg(OH)2. This creates more active surface area and results in the expedited corrosion process in the high NH4+ solutions [43,44]. With increasing immersion time, the Rct and Rf increase gradually in the 0.01 M and 0.1 M NH4+ solutions, revealing the blocking effect of the corrosion products that precipitated on the sample surface. The fluctuations of Rct and Rf in 0.001 M NH4+ solution can be ascribed to the competition between film growth and dissolution processes [41,46,60,61] on account of the slight corrosion in this environment. In addition, the variation of the capacitance values may be attributed to the variation of the film thickness and the available area. ...
2
1990
... Fig. 11 compares the corrosion rate obtained by weight loss, hydrogen evolution, and EIS after immersion for different time. A general decrease of corrosion rate in the three solutions is observed despite some fluctuations in dilute environments. The decrease of corrosion rate with increasing immersion can be ascribed to the increase of solution pH [41,42], formation of corrosion products which decreases the active surface area [[43], [44], [45]], and the decrease of NH4+ content at high pH environment [27]. The fluctuation of corrosion rate in solution with 0.001 M NH4+ is attributed to the local rupture of the surface film [10,41]. ...
... As reported by Buggio et al. [28], the microstructure of Mg alloy, the nature of the second salt or its proportion appeared to be unimportant as the anodic (or cathodic) potential was raised in NH4+-based solution. Pébère et al. [45] investigated the electrochemical behavior of magnesium using SO42- as a supporting electrolyte because of the low aggressive of SO42-. Cui et al. [58] reported that SO42- ions had no obvious effect on the icorr in saline solution. Zeng et al. [42] reported that SO42- exhibited little effect on the corrosion of magnesium alloy. Therefore, compared with the strong effect of NH4+ ion, the effect of SO42- on Mg corrosion is negligible. Potentiodynamic polarization curves of AZ31 magnesium in 0.005 M (NH4)2SO4 and Na2SO4 (pH values adjusted with H2SO4) are shown in Fig. 13 and the extracted parameters are listed in Table 4. As shown in Fig. 13, the cathodic Tafel slope of the polarization curve increases with the addition of NH4+. Moreover, there is a pseudo-passive region at the anodic branch of AZ31 in all Na2SO4 solutions. Therefore, Fig. 13 indicates that the protective MgO film of the surface could be dissolved by the NH4+. However, the influence of H+ on the corrosion behavior of AZ31 magnesium can be neglected. In view of this concentration, the effect of NH4+ on the corrosion rate of the AZ31 magnesium alloy is over 88%. Similar results are also found when comparing the polarization curves in 0.05 M (NH4)2SO4 and Na2SO4 with the same original pH (Fig. S3, in Supporting Information). ...
2
2013
... Notably, there exist dissimilarities between the corrosion rates obtained by different methods because of the intrinsic differences of these approaches. Generally, the corrosion rate calculated by hydrogen evolution is relatively lower as compared to other techniques. On one hand, the increase in the released hydrogen volume is low in the final stage and is difficult to differentiate. On the other hand, formation of hydride sub-products consumes the generated H2 and decreases the apparent corrosion rate [5,28,46]. Buggio et al. [28] revealed that the chemical recombination of hydrogen atoms of oxidic metastable hydrides was the kinetically determinant process during anodic oxidation of Mg in NH4+-containing electrolytes. In the 0.001 M NH4+ solution, corrosion rate derived from weight loss method is the highest, which may be attributed to the Non-Faraday process, including the undermining of second phases [47] and the dissolution of univalent magnesium ions [33], that cannot be detected by electrochemical methods. In 0.01 M and 0.1 M NH4+ solution, corrosion rate obtained by EIS is higher than that obtained by weight loss, especially in the 0.1 M NH4+ solution. Several reasons may be responsible for this phenomenon. Firstly, the electrochemical dissolution process may overshadow the Non-Faraday process, yielding a higher corrosion rate obtained by EIS. Secondly, EIS is an instantaneous approach, which provides a corrosion rate at a particular time over which the test is performed [8]. Therefore, this rate may be higher than the average corrosion rate that extracted from mass-loss method. ...
... Firstly, the two capacitive loops at high frequency and low frequency in all the three solutions can be attributed to the charge transfer process and the corrosion product film, respectively. Both of the charge transfer resistance and film resistance significantly decrease with increasing NH4+ concentration, suggesting the acceleration of the corrosion process. According to Cao et al. [30], the inner MgO layer was easily to be dissolved by NH4+ as compared to the Mg(OH)2. This creates more active surface area and results in the expedited corrosion process in the high NH4+ solutions [43,44]. With increasing immersion time, the Rct and Rf increase gradually in the 0.01 M and 0.1 M NH4+ solutions, revealing the blocking effect of the corrosion products that precipitated on the sample surface. The fluctuations of Rct and Rf in 0.001 M NH4+ solution can be ascribed to the competition between film growth and dissolution processes [41,46,60,61] on account of the slight corrosion in this environment. In addition, the variation of the capacitance values may be attributed to the variation of the film thickness and the available area. ...
1
2011
... Notably, there exist dissimilarities between the corrosion rates obtained by different methods because of the intrinsic differences of these approaches. Generally, the corrosion rate calculated by hydrogen evolution is relatively lower as compared to other techniques. On one hand, the increase in the released hydrogen volume is low in the final stage and is difficult to differentiate. On the other hand, formation of hydride sub-products consumes the generated H2 and decreases the apparent corrosion rate [5,28,46]. Buggio et al. [28] revealed that the chemical recombination of hydrogen atoms of oxidic metastable hydrides was the kinetically determinant process during anodic oxidation of Mg in NH4+-containing electrolytes. In the 0.001 M NH4+ solution, corrosion rate derived from weight loss method is the highest, which may be attributed to the Non-Faraday process, including the undermining of second phases [47] and the dissolution of univalent magnesium ions [33], that cannot be detected by electrochemical methods. In 0.01 M and 0.1 M NH4+ solution, corrosion rate obtained by EIS is higher than that obtained by weight loss, especially in the 0.1 M NH4+ solution. Several reasons may be responsible for this phenomenon. Firstly, the electrochemical dissolution process may overshadow the Non-Faraday process, yielding a higher corrosion rate obtained by EIS. Secondly, EIS is an instantaneous approach, which provides a corrosion rate at a particular time over which the test is performed [8]. Therefore, this rate may be higher than the average corrosion rate that extracted from mass-loss method. ...
1
1992
... Cui et al. [27] reported that the amount of NH4+ is more than 90% in a solution with a pH less than 8. Therefore, NH4+ can influence the whole corrosion process of AZ31 magnesium alloy. Firstly, the complexation of NH4+ is excluded because the complexing action between NH4+ and Mg is negligible and the Zn in AZ31 alloy is limited to cause a considerable complexation [27,48]. Even so, the presence of NH4+ may affect the surface corrosion products and expedite the corrosion processes. In Fig. 1(c), the pH values of all solutions after a 5-day immersion are below 8.4, under which the stable Mg(OH)2 layer is difficult to be preserved because the precipitation of brucite is possible when the pH exceeds 9.43 [49]. This has been confirmed by XRD and FTIR results (Fig. 4) where the peak from Mg(OH)2 is not detected. Additionally, Cao et al. [30] reported that the acceleration effect of NH4+ on Mg corrosion could be attributed to the dissolution of the thin protective inner MgO layer/outer non-protective Mg(OH)2 layer in the corrosion product film. The dissolution reactions are as follows: ...
1
2019
... Cui et al. [27] reported that the amount of NH4+ is more than 90% in a solution with a pH less than 8. Therefore, NH4+ can influence the whole corrosion process of AZ31 magnesium alloy. Firstly, the complexation of NH4+ is excluded because the complexing action between NH4+ and Mg is negligible and the Zn in AZ31 alloy is limited to cause a considerable complexation [27,48]. Even so, the presence of NH4+ may affect the surface corrosion products and expedite the corrosion processes. In Fig. 1(c), the pH values of all solutions after a 5-day immersion are below 8.4, under which the stable Mg(OH)2 layer is difficult to be preserved because the precipitation of brucite is possible when the pH exceeds 9.43 [49]. This has been confirmed by XRD and FTIR results (Fig. 4) where the peak from Mg(OH)2 is not detected. Additionally, Cao et al. [30] reported that the acceleration effect of NH4+ on Mg corrosion could be attributed to the dissolution of the thin protective inner MgO layer/outer non-protective Mg(OH)2 layer in the corrosion product film. The dissolution reactions are as follows: ...
1
2009
... Moreover, ammonium ion preferentially degrades MgO, which is assumed to be more protective than Mg(OH)2 [30,43,50]. Actually, there must be a thin Mg(OH)2 and MgO film formed at the interface of solution and magnesium, which may be caused by the partial alkalization on account of the cathodic reactions during dissolution of magnesium [51]. Due to the dissolution of the corrosion product layer, a fresh surface of AZ31 magnesium alloy is exposed, and the substrate can directly react with the solution without any barrier. Eventually, the active surface area of AZ31 magnesium alloy decreases and the bare surface is covered by a loose corrosion product film, which may be constituted by various ions through a weak-interaction. ...
1
2008
... Moreover, ammonium ion preferentially degrades MgO, which is assumed to be more protective than Mg(OH)2 [30,43,50]. Actually, there must be a thin Mg(OH)2 and MgO film formed at the interface of solution and magnesium, which may be caused by the partial alkalization on account of the cathodic reactions during dissolution of magnesium [51]. Due to the dissolution of the corrosion product layer, a fresh surface of AZ31 magnesium alloy is exposed, and the substrate can directly react with the solution without any barrier. Eventually, the active surface area of AZ31 magnesium alloy decreases and the bare surface is covered by a loose corrosion product film, which may be constituted by various ions through a weak-interaction. ...
1
2009
... XRD analysis implies the presence of Mg-Al-CO32- hydrotalcite-like compound (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O (HTLc) and Mg5(CO3)4(OH)2·5H2O (Section 3.2). The presence of Mg5(CO3)4(OH)2·5H2O is likely attributed to the diffusion of CO2 from the environment, which reacts with unstable Mg(OH)2 [52,53]. Arrabal et al. [54,55] found the presence of (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O on the magnesium alloy after immersion in 3.5 wt.% NaCl. Jang et al. [56] also found that the (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O was the primary corrosion product on AZ31 alloy in simulated physiological solutions. In atmospheric environment, Liao et al. [13] demonstrated the existence of the HTLc as the dominant corrosion products and proposed a simplified formation process. They considered that the high temperature on sunny days and the high local pH promoted the dissolution of Al3+ ions and thus the (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O was developed. In the present work, the solution is acidic in the initial immersion period in the presence of NH4+. This could accelerate the dissolution of Al to Al3+ and enhance the formation of (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O in the later immersion stage, during which the formation of Al-rich precursors of hydrotalcite (HT) and exchange of OH- for CO32- are feasible [57]. This HT-like compound is similar to HT and presents a structure related to brucite with the general formula [Mg1-x2+Mx3+(OH)2]x+Ax/nn-·mH2O, where M2+ and M3+ are divalent and trivalent metals, respectively, and An- is an anion [13,57]. ...
1
2017
... XRD analysis implies the presence of Mg-Al-CO32- hydrotalcite-like compound (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O (HTLc) and Mg5(CO3)4(OH)2·5H2O (Section 3.2). The presence of Mg5(CO3)4(OH)2·5H2O is likely attributed to the diffusion of CO2 from the environment, which reacts with unstable Mg(OH)2 [52,53]. Arrabal et al. [54,55] found the presence of (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O on the magnesium alloy after immersion in 3.5 wt.% NaCl. Jang et al. [56] also found that the (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O was the primary corrosion product on AZ31 alloy in simulated physiological solutions. In atmospheric environment, Liao et al. [13] demonstrated the existence of the HTLc as the dominant corrosion products and proposed a simplified formation process. They considered that the high temperature on sunny days and the high local pH promoted the dissolution of Al3+ ions and thus the (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O was developed. In the present work, the solution is acidic in the initial immersion period in the presence of NH4+. This could accelerate the dissolution of Al to Al3+ and enhance the formation of (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O in the later immersion stage, during which the formation of Al-rich precursors of hydrotalcite (HT) and exchange of OH- for CO32- are feasible [57]. This HT-like compound is similar to HT and presents a structure related to brucite with the general formula [Mg1-x2+Mx3+(OH)2]x+Ax/nn-·mH2O, where M2+ and M3+ are divalent and trivalent metals, respectively, and An- is an anion [13,57]. ...
1
2012
... XRD analysis implies the presence of Mg-Al-CO32- hydrotalcite-like compound (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O (HTLc) and Mg5(CO3)4(OH)2·5H2O (Section 3.2). The presence of Mg5(CO3)4(OH)2·5H2O is likely attributed to the diffusion of CO2 from the environment, which reacts with unstable Mg(OH)2 [52,53]. Arrabal et al. [54,55] found the presence of (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O on the magnesium alloy after immersion in 3.5 wt.% NaCl. Jang et al. [56] also found that the (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O was the primary corrosion product on AZ31 alloy in simulated physiological solutions. In atmospheric environment, Liao et al. [13] demonstrated the existence of the HTLc as the dominant corrosion products and proposed a simplified formation process. They considered that the high temperature on sunny days and the high local pH promoted the dissolution of Al3+ ions and thus the (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O was developed. In the present work, the solution is acidic in the initial immersion period in the presence of NH4+. This could accelerate the dissolution of Al to Al3+ and enhance the formation of (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O in the later immersion stage, during which the formation of Al-rich precursors of hydrotalcite (HT) and exchange of OH- for CO32- are feasible [57]. This HT-like compound is similar to HT and presents a structure related to brucite with the general formula [Mg1-x2+Mx3+(OH)2]x+Ax/nn-·mH2O, where M2+ and M3+ are divalent and trivalent metals, respectively, and An- is an anion [13,57]. ...
1
2012
... XRD analysis implies the presence of Mg-Al-CO32- hydrotalcite-like compound (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O (HTLc) and Mg5(CO3)4(OH)2·5H2O (Section 3.2). The presence of Mg5(CO3)4(OH)2·5H2O is likely attributed to the diffusion of CO2 from the environment, which reacts with unstable Mg(OH)2 [52,53]. Arrabal et al. [54,55] found the presence of (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O on the magnesium alloy after immersion in 3.5 wt.% NaCl. Jang et al. [56] also found that the (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O was the primary corrosion product on AZ31 alloy in simulated physiological solutions. In atmospheric environment, Liao et al. [13] demonstrated the existence of the HTLc as the dominant corrosion products and proposed a simplified formation process. They considered that the high temperature on sunny days and the high local pH promoted the dissolution of Al3+ ions and thus the (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O was developed. In the present work, the solution is acidic in the initial immersion period in the presence of NH4+. This could accelerate the dissolution of Al to Al3+ and enhance the formation of (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O in the later immersion stage, during which the formation of Al-rich precursors of hydrotalcite (HT) and exchange of OH- for CO32- are feasible [57]. This HT-like compound is similar to HT and presents a structure related to brucite with the general formula [Mg1-x2+Mx3+(OH)2]x+Ax/nn-·mH2O, where M2+ and M3+ are divalent and trivalent metals, respectively, and An- is an anion [13,57]. ...
1
2013
... XRD analysis implies the presence of Mg-Al-CO32- hydrotalcite-like compound (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O (HTLc) and Mg5(CO3)4(OH)2·5H2O (Section 3.2). The presence of Mg5(CO3)4(OH)2·5H2O is likely attributed to the diffusion of CO2 from the environment, which reacts with unstable Mg(OH)2 [52,53]. Arrabal et al. [54,55] found the presence of (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O on the magnesium alloy after immersion in 3.5 wt.% NaCl. Jang et al. [56] also found that the (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O was the primary corrosion product on AZ31 alloy in simulated physiological solutions. In atmospheric environment, Liao et al. [13] demonstrated the existence of the HTLc as the dominant corrosion products and proposed a simplified formation process. They considered that the high temperature on sunny days and the high local pH promoted the dissolution of Al3+ ions and thus the (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O was developed. In the present work, the solution is acidic in the initial immersion period in the presence of NH4+. This could accelerate the dissolution of Al to Al3+ and enhance the formation of (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O in the later immersion stage, during which the formation of Al-rich precursors of hydrotalcite (HT) and exchange of OH- for CO32- are feasible [57]. This HT-like compound is similar to HT and presents a structure related to brucite with the general formula [Mg1-x2+Mx3+(OH)2]x+Ax/nn-·mH2O, where M2+ and M3+ are divalent and trivalent metals, respectively, and An- is an anion [13,57]. ...
2
2012
... XRD analysis implies the presence of Mg-Al-CO32- hydrotalcite-like compound (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O (HTLc) and Mg5(CO3)4(OH)2·5H2O (Section 3.2). The presence of Mg5(CO3)4(OH)2·5H2O is likely attributed to the diffusion of CO2 from the environment, which reacts with unstable Mg(OH)2 [52,53]. Arrabal et al. [54,55] found the presence of (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O on the magnesium alloy after immersion in 3.5 wt.% NaCl. Jang et al. [56] also found that the (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O was the primary corrosion product on AZ31 alloy in simulated physiological solutions. In atmospheric environment, Liao et al. [13] demonstrated the existence of the HTLc as the dominant corrosion products and proposed a simplified formation process. They considered that the high temperature on sunny days and the high local pH promoted the dissolution of Al3+ ions and thus the (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O was developed. In the present work, the solution is acidic in the initial immersion period in the presence of NH4+. This could accelerate the dissolution of Al to Al3+ and enhance the formation of (Mg0.833Al0.167)(OH)2(CO3)0.083·0.75H2O in the later immersion stage, during which the formation of Al-rich precursors of hydrotalcite (HT) and exchange of OH- for CO32- are feasible [57]. This HT-like compound is similar to HT and presents a structure related to brucite with the general formula [Mg1-x2+Mx3+(OH)2]x+Ax/nn-·mH2O, where M2+ and M3+ are divalent and trivalent metals, respectively, and An- is an anion [13,57]. ...
... ,57]. ...
1
2017
... As reported by Buggio et al. [28], the microstructure of Mg alloy, the nature of the second salt or its proportion appeared to be unimportant as the anodic (or cathodic) potential was raised in NH4+-based solution. Pébère et al. [45] investigated the electrochemical behavior of magnesium using SO42- as a supporting electrolyte because of the low aggressive of SO42-. Cui et al. [58] reported that SO42- ions had no obvious effect on the icorr in saline solution. Zeng et al. [42] reported that SO42- exhibited little effect on the corrosion of magnesium alloy. Therefore, compared with the strong effect of NH4+ ion, the effect of SO42- on Mg corrosion is negligible. Potentiodynamic polarization curves of AZ31 magnesium in 0.005 M (NH4)2SO4 and Na2SO4 (pH values adjusted with H2SO4) are shown in Fig. 13 and the extracted parameters are listed in Table 4. As shown in Fig. 13, the cathodic Tafel slope of the polarization curve increases with the addition of NH4+. Moreover, there is a pseudo-passive region at the anodic branch of AZ31 in all Na2SO4 solutions. Therefore, Fig. 13 indicates that the protective MgO film of the surface could be dissolved by the NH4+. However, the influence of H+ on the corrosion behavior of AZ31 magnesium can be neglected. In view of this concentration, the effect of NH4+ on the corrosion rate of the AZ31 magnesium alloy is over 88%. Similar results are also found when comparing the polarization curves in 0.05 M (NH4)2SO4 and Na2SO4 with the same original pH (Fig. S3, in Supporting Information). ...
2
2019
... The interfacial process occurring on the magnesium/solution interface varies with NH4+ concentration as indicated by the change of EIS loops. To intuitively present the evolution of EIS data with increasing immersion time and NH4+ concentration, the normalized impedance diagrams which are proposed by Baril et al. [43] are plotted and shown in Fig. 14. The diagrams measured in 0.001 M NH4+ solution coincide well with each other, indicating the same corrosion mechanism during the immersion test [59]. In 0.01 M NH4+ solution, the inductive loops shrink with increasing immersion time, while an additional inductive loop is detected at low frequency in the 0.1 M NH4+ solution, suggesting that aging in the solution affects the interfacial processes. Moreover, the diagrams at 0.5 h in the three solutions show distinct characteristics, revealing that NH4+ concentration also influence the dissolution process occurring on the magnesium/solution interface. ...
... Secondly, inductive loops appear in the EIS diagrams of AZ31 magnesium alloys in 0.01 M and 0.1 M NH4+ solutions, with the latter exhibiting two loops during the initial immersion periods. Several mechanisms have been proposed to interpret the inductance loops in EIS spectra of magnesium alloys in the literatures [30,37,59,[62], [63], [64], [65]], including the Mg+ theory, the adsorption/desorption of intermediate species, and the initiation of localized corrosion. As shown in Fig. 3, the distinct difference between the surface layer morphology in 0.001 M NH4+ and the other solutions is the relatively intact surface film without obvious spalling (Fig. 3(a2) and (a3)). Therefore, the inductive loops in 0.01 M and 0.1 M NH4+ may be attributed to the local destruction of surface layer which contributes to the occurrence of localized corrosion and the adsorption of intermediate species. This agrees well with Gomes et al. [59] and Chen et al. [66] who found the similar inductive loops in the sulfate-based solutions. The other inductive loop in the lowest frequency range in 0.1 M NH4+ solution may be attributed to the adsorption of hydrogen atoms. In this electrolyte, the prominent characteristic is the severe hydrogen evolution during the initial immersion period (Fig. 2(a)). Because hydrogen evolution reaction can be divided into two procedures including the electrochemical dissolution and electrochemical desorption, the lower rate of the latter one creates considerable Hads on the sample surface. According to Cao et al. [67] and Armstrong et al. [68], this intermediate product yields an inductive loop which can reasonably explains the particular inductive loop in the most concentrated solution during the initial immersion stage. ...
1
2020
... Firstly, the two capacitive loops at high frequency and low frequency in all the three solutions can be attributed to the charge transfer process and the corrosion product film, respectively. Both of the charge transfer resistance and film resistance significantly decrease with increasing NH4+ concentration, suggesting the acceleration of the corrosion process. According to Cao et al. [30], the inner MgO layer was easily to be dissolved by NH4+ as compared to the Mg(OH)2. This creates more active surface area and results in the expedited corrosion process in the high NH4+ solutions [43,44]. With increasing immersion time, the Rct and Rf increase gradually in the 0.01 M and 0.1 M NH4+ solutions, revealing the blocking effect of the corrosion products that precipitated on the sample surface. The fluctuations of Rct and Rf in 0.001 M NH4+ solution can be ascribed to the competition between film growth and dissolution processes [41,46,60,61] on account of the slight corrosion in this environment. In addition, the variation of the capacitance values may be attributed to the variation of the film thickness and the available area. ...
1
2019
... Firstly, the two capacitive loops at high frequency and low frequency in all the three solutions can be attributed to the charge transfer process and the corrosion product film, respectively. Both of the charge transfer resistance and film resistance significantly decrease with increasing NH4+ concentration, suggesting the acceleration of the corrosion process. According to Cao et al. [30], the inner MgO layer was easily to be dissolved by NH4+ as compared to the Mg(OH)2. This creates more active surface area and results in the expedited corrosion process in the high NH4+ solutions [43,44]. With increasing immersion time, the Rct and Rf increase gradually in the 0.01 M and 0.1 M NH4+ solutions, revealing the blocking effect of the corrosion products that precipitated on the sample surface. The fluctuations of Rct and Rf in 0.001 M NH4+ solution can be ascribed to the competition between film growth and dissolution processes [41,46,60,61] on account of the slight corrosion in this environment. In addition, the variation of the capacitance values may be attributed to the variation of the film thickness and the available area. ...
1
2006
... Secondly, inductive loops appear in the EIS diagrams of AZ31 magnesium alloys in 0.01 M and 0.1 M NH4+ solutions, with the latter exhibiting two loops during the initial immersion periods. Several mechanisms have been proposed to interpret the inductance loops in EIS spectra of magnesium alloys in the literatures [30,37,59,[62], [63], [64], [65]], including the Mg+ theory, the adsorption/desorption of intermediate species, and the initiation of localized corrosion. As shown in Fig. 3, the distinct difference between the surface layer morphology in 0.001 M NH4+ and the other solutions is the relatively intact surface film without obvious spalling (Fig. 3(a2) and (a3)). Therefore, the inductive loops in 0.01 M and 0.1 M NH4+ may be attributed to the local destruction of surface layer which contributes to the occurrence of localized corrosion and the adsorption of intermediate species. This agrees well with Gomes et al. [59] and Chen et al. [66] who found the similar inductive loops in the sulfate-based solutions. The other inductive loop in the lowest frequency range in 0.1 M NH4+ solution may be attributed to the adsorption of hydrogen atoms. In this electrolyte, the prominent characteristic is the severe hydrogen evolution during the initial immersion period (Fig. 2(a)). Because hydrogen evolution reaction can be divided into two procedures including the electrochemical dissolution and electrochemical desorption, the lower rate of the latter one creates considerable Hads on the sample surface. According to Cao et al. [67] and Armstrong et al. [68], this intermediate product yields an inductive loop which can reasonably explains the particular inductive loop in the most concentrated solution during the initial immersion stage. ...
1
2016
... Secondly, inductive loops appear in the EIS diagrams of AZ31 magnesium alloys in 0.01 M and 0.1 M NH4+ solutions, with the latter exhibiting two loops during the initial immersion periods. Several mechanisms have been proposed to interpret the inductance loops in EIS spectra of magnesium alloys in the literatures [30,37,59,[62], [63], [64], [65]], including the Mg+ theory, the adsorption/desorption of intermediate species, and the initiation of localized corrosion. As shown in Fig. 3, the distinct difference between the surface layer morphology in 0.001 M NH4+ and the other solutions is the relatively intact surface film without obvious spalling (Fig. 3(a2) and (a3)). Therefore, the inductive loops in 0.01 M and 0.1 M NH4+ may be attributed to the local destruction of surface layer which contributes to the occurrence of localized corrosion and the adsorption of intermediate species. This agrees well with Gomes et al. [59] and Chen et al. [66] who found the similar inductive loops in the sulfate-based solutions. The other inductive loop in the lowest frequency range in 0.1 M NH4+ solution may be attributed to the adsorption of hydrogen atoms. In this electrolyte, the prominent characteristic is the severe hydrogen evolution during the initial immersion period (Fig. 2(a)). Because hydrogen evolution reaction can be divided into two procedures including the electrochemical dissolution and electrochemical desorption, the lower rate of the latter one creates considerable Hads on the sample surface. According to Cao et al. [67] and Armstrong et al. [68], this intermediate product yields an inductive loop which can reasonably explains the particular inductive loop in the most concentrated solution during the initial immersion stage. ...
1
2020
... Secondly, inductive loops appear in the EIS diagrams of AZ31 magnesium alloys in 0.01 M and 0.1 M NH4+ solutions, with the latter exhibiting two loops during the initial immersion periods. Several mechanisms have been proposed to interpret the inductance loops in EIS spectra of magnesium alloys in the literatures [30,37,59,[62], [63], [64], [65]], including the Mg+ theory, the adsorption/desorption of intermediate species, and the initiation of localized corrosion. As shown in Fig. 3, the distinct difference between the surface layer morphology in 0.001 M NH4+ and the other solutions is the relatively intact surface film without obvious spalling (Fig. 3(a2) and (a3)). Therefore, the inductive loops in 0.01 M and 0.1 M NH4+ may be attributed to the local destruction of surface layer which contributes to the occurrence of localized corrosion and the adsorption of intermediate species. This agrees well with Gomes et al. [59] and Chen et al. [66] who found the similar inductive loops in the sulfate-based solutions. The other inductive loop in the lowest frequency range in 0.1 M NH4+ solution may be attributed to the adsorption of hydrogen atoms. In this electrolyte, the prominent characteristic is the severe hydrogen evolution during the initial immersion period (Fig. 2(a)). Because hydrogen evolution reaction can be divided into two procedures including the electrochemical dissolution and electrochemical desorption, the lower rate of the latter one creates considerable Hads on the sample surface. According to Cao et al. [67] and Armstrong et al. [68], this intermediate product yields an inductive loop which can reasonably explains the particular inductive loop in the most concentrated solution during the initial immersion stage. ...
1
2015
... Secondly, inductive loops appear in the EIS diagrams of AZ31 magnesium alloys in 0.01 M and 0.1 M NH4+ solutions, with the latter exhibiting two loops during the initial immersion periods. Several mechanisms have been proposed to interpret the inductance loops in EIS spectra of magnesium alloys in the literatures [30,37,59,[62], [63], [64], [65]], including the Mg+ theory, the adsorption/desorption of intermediate species, and the initiation of localized corrosion. As shown in Fig. 3, the distinct difference between the surface layer morphology in 0.001 M NH4+ and the other solutions is the relatively intact surface film without obvious spalling (Fig. 3(a2) and (a3)). Therefore, the inductive loops in 0.01 M and 0.1 M NH4+ may be attributed to the local destruction of surface layer which contributes to the occurrence of localized corrosion and the adsorption of intermediate species. This agrees well with Gomes et al. [59] and Chen et al. [66] who found the similar inductive loops in the sulfate-based solutions. The other inductive loop in the lowest frequency range in 0.1 M NH4+ solution may be attributed to the adsorption of hydrogen atoms. In this electrolyte, the prominent characteristic is the severe hydrogen evolution during the initial immersion period (Fig. 2(a)). Because hydrogen evolution reaction can be divided into two procedures including the electrochemical dissolution and electrochemical desorption, the lower rate of the latter one creates considerable Hads on the sample surface. According to Cao et al. [67] and Armstrong et al. [68], this intermediate product yields an inductive loop which can reasonably explains the particular inductive loop in the most concentrated solution during the initial immersion stage. ...
1
2007
... Secondly, inductive loops appear in the EIS diagrams of AZ31 magnesium alloys in 0.01 M and 0.1 M NH4+ solutions, with the latter exhibiting two loops during the initial immersion periods. Several mechanisms have been proposed to interpret the inductance loops in EIS spectra of magnesium alloys in the literatures [30,37,59,[62], [63], [64], [65]], including the Mg+ theory, the adsorption/desorption of intermediate species, and the initiation of localized corrosion. As shown in Fig. 3, the distinct difference between the surface layer morphology in 0.001 M NH4+ and the other solutions is the relatively intact surface film without obvious spalling (Fig. 3(a2) and (a3)). Therefore, the inductive loops in 0.01 M and 0.1 M NH4+ may be attributed to the local destruction of surface layer which contributes to the occurrence of localized corrosion and the adsorption of intermediate species. This agrees well with Gomes et al. [59] and Chen et al. [66] who found the similar inductive loops in the sulfate-based solutions. The other inductive loop in the lowest frequency range in 0.1 M NH4+ solution may be attributed to the adsorption of hydrogen atoms. In this electrolyte, the prominent characteristic is the severe hydrogen evolution during the initial immersion period (Fig. 2(a)). Because hydrogen evolution reaction can be divided into two procedures including the electrochemical dissolution and electrochemical desorption, the lower rate of the latter one creates considerable Hads on the sample surface. According to Cao et al. [67] and Armstrong et al. [68], this intermediate product yields an inductive loop which can reasonably explains the particular inductive loop in the most concentrated solution during the initial immersion stage. ...
1
1990
... Secondly, inductive loops appear in the EIS diagrams of AZ31 magnesium alloys in 0.01 M and 0.1 M NH4+ solutions, with the latter exhibiting two loops during the initial immersion periods. Several mechanisms have been proposed to interpret the inductance loops in EIS spectra of magnesium alloys in the literatures [30,37,59,[62], [63], [64], [65]], including the Mg+ theory, the adsorption/desorption of intermediate species, and the initiation of localized corrosion. As shown in Fig. 3, the distinct difference between the surface layer morphology in 0.001 M NH4+ and the other solutions is the relatively intact surface film without obvious spalling (Fig. 3(a2) and (a3)). Therefore, the inductive loops in 0.01 M and 0.1 M NH4+ may be attributed to the local destruction of surface layer which contributes to the occurrence of localized corrosion and the adsorption of intermediate species. This agrees well with Gomes et al. [59] and Chen et al. [66] who found the similar inductive loops in the sulfate-based solutions. The other inductive loop in the lowest frequency range in 0.1 M NH4+ solution may be attributed to the adsorption of hydrogen atoms. In this electrolyte, the prominent characteristic is the severe hydrogen evolution during the initial immersion period (Fig. 2(a)). Because hydrogen evolution reaction can be divided into two procedures including the electrochemical dissolution and electrochemical desorption, the lower rate of the latter one creates considerable Hads on the sample surface. According to Cao et al. [67] and Armstrong et al. [68], this intermediate product yields an inductive loop which can reasonably explains the particular inductive loop in the most concentrated solution during the initial immersion stage. ...
1
1972
... Secondly, inductive loops appear in the EIS diagrams of AZ31 magnesium alloys in 0.01 M and 0.1 M NH4+ solutions, with the latter exhibiting two loops during the initial immersion periods. Several mechanisms have been proposed to interpret the inductance loops in EIS spectra of magnesium alloys in the literatures [30,37,59,[62], [63], [64], [65]], including the Mg+ theory, the adsorption/desorption of intermediate species, and the initiation of localized corrosion. As shown in Fig. 3, the distinct difference between the surface layer morphology in 0.001 M NH4+ and the other solutions is the relatively intact surface film without obvious spalling (Fig. 3(a2) and (a3)). Therefore, the inductive loops in 0.01 M and 0.1 M NH4+ may be attributed to the local destruction of surface layer which contributes to the occurrence of localized corrosion and the adsorption of intermediate species. This agrees well with Gomes et al. [59] and Chen et al. [66] who found the similar inductive loops in the sulfate-based solutions. The other inductive loop in the lowest frequency range in 0.1 M NH4+ solution may be attributed to the adsorption of hydrogen atoms. In this electrolyte, the prominent characteristic is the severe hydrogen evolution during the initial immersion period (Fig. 2(a)). Because hydrogen evolution reaction can be divided into two procedures including the electrochemical dissolution and electrochemical desorption, the lower rate of the latter one creates considerable Hads on the sample surface. According to Cao et al. [67] and Armstrong et al. [68], this intermediate product yields an inductive loop which can reasonably explains the particular inductive loop in the most concentrated solution during the initial immersion stage. ...


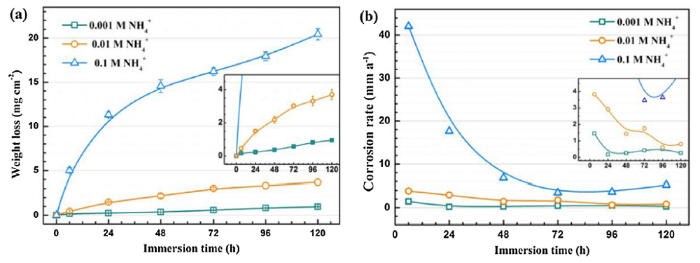
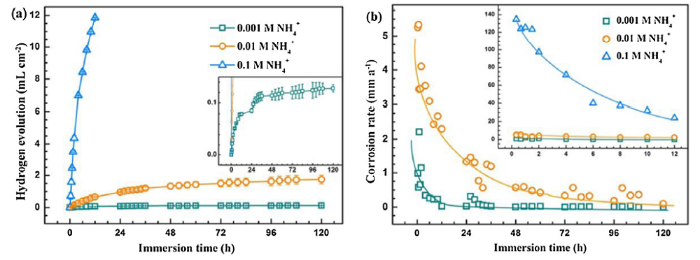

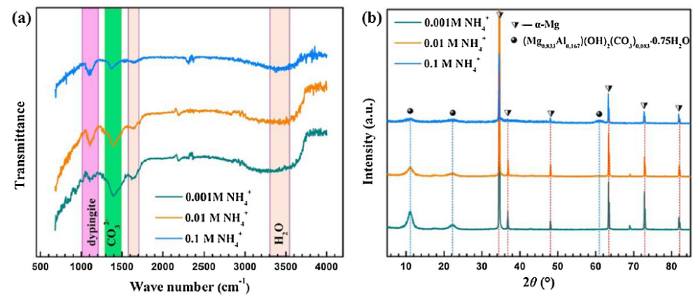


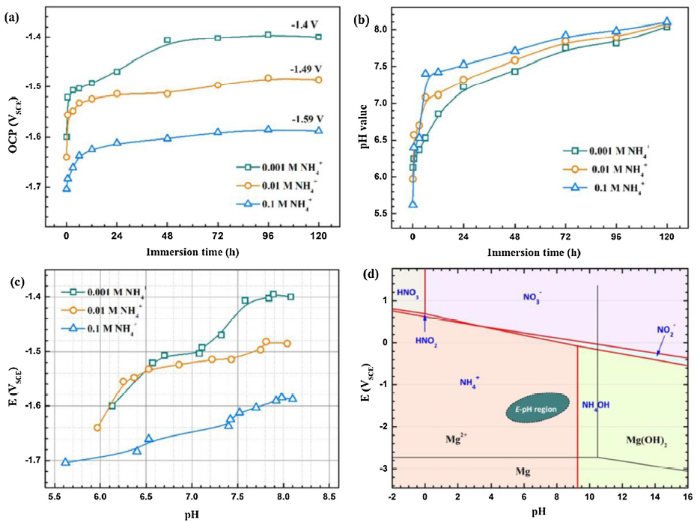
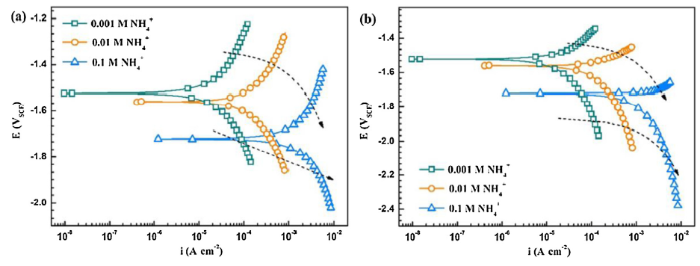
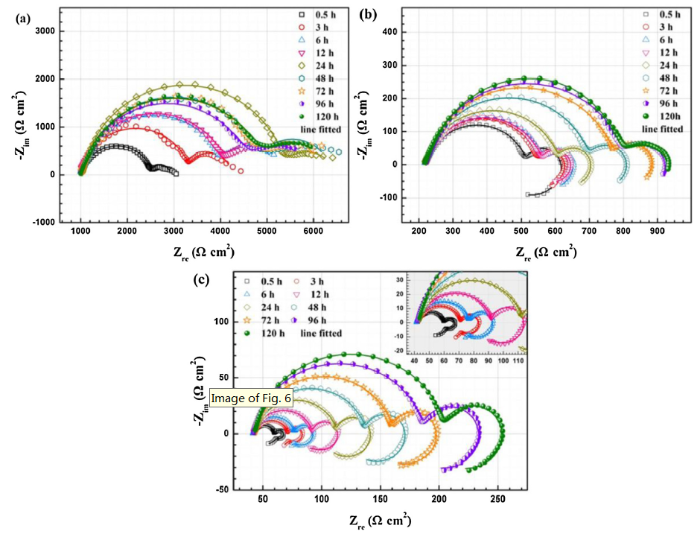
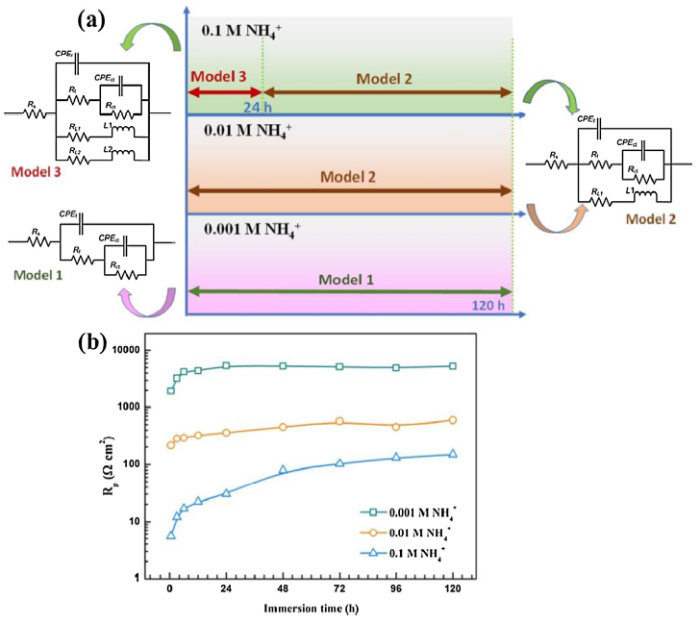
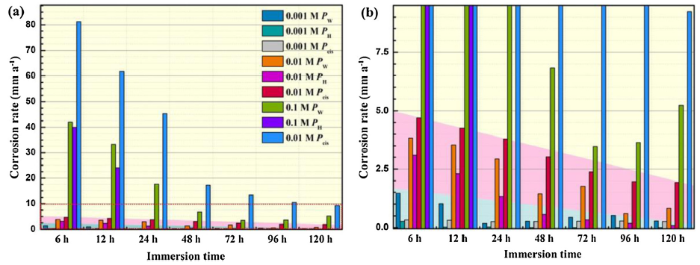
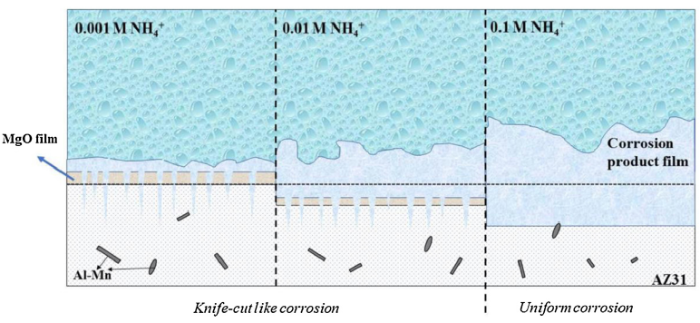
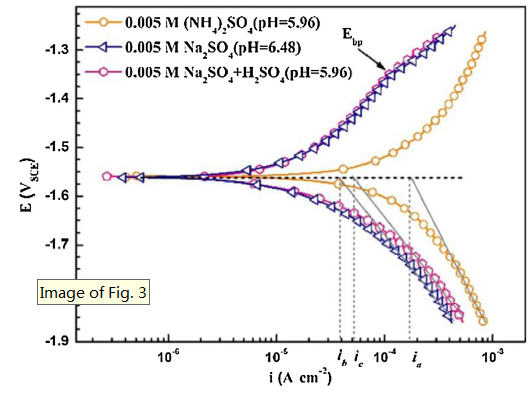



 WeChat
WeChat
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}