Machine learning in materials genome initiative: A review
Yingli Liua,b, Chen Niua, Zhuo Wang,c,f,a,*,1, Yong Gand, Yan Zhua, Shuhong Sune, Tao Shen,a,b,**,1
aFaculty of Information Engineering and Automation, Kunming University of Science and Technology, Kunming, 650500, China
bComputer Technology Application Key Lab of Yunnan Province, Kunming University of Science and Technology, Kunming, 650500, China
cLight Alloy Research Institute, Central South University, Changsha, 410083, China
dChinese Academy of Engineering, Beijing, 100088, China
eFaculty of Materials Science and Engineering, Kunming University of Science and Technology, Kunming, 650093, China
fChengdu MatAi Technology Co., Ltd, Chengdu, 610041, China
Corresponding authors:∗ Light Alloy Research Institute, Central South University, Changsha, 410083, China.wangzhuo@mat.ai(Z. Wang),∗∗ Faculty of Information Engineering and Automation, Kunming University of Science and Technology, Kunming, 650500, China. E-mail addresses:shentao@kust.edu.cn(T. Shen).
Discovering new materials with excellent performance is a hot issue in the materials genome initiative. Traditional experiments and calculations often waste large amounts of time and money and are also limited by various conditions. Therefore, it is imperative to develop a new method to accelerate the discovery and design of new materials. In recent years, material discovery and design methods using machine learning have attracted much attention from material experts and have made some progress. This review first outlines available materials database and material data analytics tools and then elaborates on the machine learning algorithms used in materials science. Next, the field of application of machine learning in materials science is summarized, focusing on the aspects of structure determination, performance prediction, fingerprint prediction, and new material discovery. Finally, the review points out the problems of data and machine learning in materials science and points to future research. Using machine learning algorithms, the authors hope to achieve amazing results in material discovery and design.
Yingli Liu, Chen Niu, Zhuo Wang, Yong Gan, Yan Zhu, Shuhong Sun, Tao Shen. Machine learning in materials genome initiative: A review. Journal of Materials Science & Technology[J], 2020, 57(0): 113-122 DOI:10.1016/j.jmst.2020.01.067
1. Introduction
At the beginning of the 21st Century, Ceder, who worked at the Massachusetts Institute of Technology (MIT; Cambridge, MA, USA) was influenced by high-throughput, data-driven material discovery methods and inspired by the Human Genome Project. He considered whether material scientists could learn from the experience of geneticists. In 2006, Ceder launched the Materials Genomics project at the MIT, using an improved data-mining algorithm to predict lithium-based materials for electric vehicle batteries. By 2010, the project had evolved to include approximately 20,000 predicted compounds [1]. At the same time, at Duke University (Durham, NC, USA), Curtarolo established the Materials Genomics Center, focusing on metal alloy research. His team gradually extended the 2003 algorithm and library [2] to AFLOW (Automatic Flow), a system that can calculate known crystal structures and automatically predict new structures [3]. Despite this, computational materials science was still not valued by experts until the American White House announced hundreds of millions of dollars of funding for the Materials Genome Initiative (MGI) in June 2011 [1].
As highlighted by the MGI, theory and simulation can enhance the efficiency of materials discovery and design. On the other hand, a purely theoretical and simulation approach to materials discovery and design is often tricky to realize experimentally due to complications in synthesis, existence, and production of defects and possible metastable states. To bridge this gap, all aspects of theory and simulation must be tightly integrated with experiments and data [4]. High-throughput density functional theory has become a universal tool for material discovery [[5], [6], [7]], but its limitation is that computational costs are high [8]. The need now is to overcome this limitation to take advantage of the latest advances in data and information science [9].
Machine learning [10] is an interdisciplinary subject lying between computer science and statistics. It is the core of artificial intelligence and data science. It promotes the availability of online data and low-cost computing through algorithmic learning [11]. Even though machine learning is a rapidly innovative technology, some algorithms in machine learning are bound to be outdated, but the importance of using scientific reasoning to find reliable structure, property, and processing models will never be outdated [12]. The types of materials are diverse, and the factors affecting the results are complex. Therefore, the data relationship needs to be clear, and the machine learning method is good at discovering and establishing connections among numerous data points. The introduction of machine learning methods is beneficial in understanding and discovering the correlation between experimental parameters and materials performance.
The main processes of machine learning may be divided into data preparation, descriptor selection, algorithm/model selection, model prediction, and model application [13]. By applying this process to material discovery and design, a complete cycle is fulfilled, from experimental process data collection to performance prediction and finally to experimental validation. Machine learning methods provide an efficient toolset for extracting important correlations among material phenomena [14]. Machine learning can classify viral genomes [15] and identify type 2 diabetes [16]. In the field of chemoinformatics, machine learning models can detect outlier samples [17] and analyze chemical data [18]. It can even perform classification of potato bruise levels [19]. It is evident that machine learning methods have attracted the attention of researchers in many fields. The approach has been widely used in biology, medicine, and other research fields. In the field of materials science, some materials researchers have used machine learning methods to predict material properties.
Recently, material discovery and design using machine learning have received increasing attention and have been greatly improved from the standpoints of time efficiency and prediction accuracy [20,21]. Voyles [22] used machine learning algorithms to improve the quality of material microscope data and extract material information from enhanced data. Raccuglia et al. [23] used the support vector machine (SVM) approach to predict the feasibility of untested responses to data collected in failed experiments. Wicker et al. [24] used the SVM algorithm with an RBF kernel to predict the crystallinity of molecular materials. Rupp [25] found that combining quantum mechanics (QM) with machine learning could be expected to improve the accuracy of QM. Stanev et al. [26] proposed that a machine learning model could simulate the critical temperature of superconductors. A neural network can represent high-dimensional potential energy surfaces when the atomistic materials are simulated, and machine-learned interpolation of atomic potential energy surfaces can automatically construct high-precision atomic interaction potentials [[27], [28], [29]]. The predictive power of machine learning algorithms was also reflected in many aspects of material properties, such as sintered density [30], atomization energies [31], band gaps [32,33], and phase transitions [34,35].
This review discusses the materials database, the material data analytics tools, the machine learning methods of the material genomic data section, and application fields, as well as common problems.
2. Materials database
Continuous advancement in science depends on shared and repeatable data [36]. Large-scale data analysis is the driving force for material progress. Materials science is on the verge of adopting Materials 4.0 /fourth paradigm [37,38] or data-driven [39] discovery on large-scale data [40]. The quality of data analysis and data mining is affected by the data quality of the materials database and directly affects the application of data that are mined. The existing database is far from including all known materials, not to mention all possible materials. Data-driven discovery applies to certain materials, but not to all materials. Even if people pick an interesting material on their computers, it will take years to synthesize it in the laboratory [1].
Data can come from calculations [41], literature data [42], hypotheses, experiments, and even from failed experiments [23]. Besides, new data can also generate by interpolation of existing data [43]. The amount of material science data is not particularly large, but it is challenging to explore the diversity of data types and the complexity of objects [44].
2.1. Current main materials database
An early database, The Inorganic Crystal Structure Database [45], created in 2002, which covers the literature from 1915 to 2002, is a comprehensive collection of crystal structure information for non-organic compounds, including inorganics, minerals, ceramics, and metals [46]. In 2015, Wolverton [47] launched the Open Quantum Materials Database (OQMD), which is also an inorganic crystal structure database with approximately 50,000 pieces of data from a widely used experimental library [1]. Cambridge Structural Databases (CSD) [48] is a worldwide repository for small-molecule organic and metal-organic crystal structures hosted by Cambridge Crystallographic Data Center (CCDC) (https://www.lib.ncsu.edu/databases/cambridge-structural.html [49]). The Materials Projec t [50,51] is the core program of the Materials Genome Initiative, which uses high-throughput calculations to reveal the properties of all known inorganic materials. These open datasets can be accessed through multiple channels for interactive exploration and data mining. The Materials Project URL is https://www.materialsproject.org [52]. By September 2019, the database contained 120,612 inorganic compounds, 52,366 band structures, 35,336 molecules, 530,243 nanoporous materials, 13,621 elastic tensors, 3003 piezoelectric tensors, 4401 intercalation electrodes, and 16,128 conversion electrodes.
In 2003, Ceder et al. [1] first demonstrated how quantum mechanical computing databases could help predict the most likely crystal structure of metal alloys—a crucial step in the invention of new materials. Ceder introduced the concept of public libraries of material properties and used data mining to fill in missing data. In the second year after the genome project was launched, Curtarolo [53] posted a database based on his software, called AFLOWlib, which is a database with more than one million materials and approximately 100 million calculated properties. AFLOWlib contains such a large amount of data because it also includes thousands of hypothetical materials, many of which exist for only one second in the real world, but Curtarolo used these data to study why some alloys can form metallic glass [54]. OQMD is also a high-throughput database that includes a large number of hypothetical materials, and the entire database can be obtained for free at http://www.oqmd.org/download [47].
The maturity of high-throughput first-principles calculations, along with the rise of virtual material screening, has created many accessible databases for the materials science community, including Citrination Platform [55], large molecular libraries [56], and the Harvard Clean Energy Project Database (CEPDB) [57].
MatWeb is the world's largest database of material properties, with over 130,000 metals, plastics, ceramics, and composites [58]. Total Materia is the world’s most comprehensive materials database, which is based on 74 international standards and offers more than 15,000,000 materials [59]. ASM International is the world’s largest and most established materials information society, which can connect anyone to a global network of peers and provides access to trusted materials information [60].
In addition, there are the computational materials repository [61], computational materials property databases (www.synthesisproject.org) [42], MatNavi materials databases [62], the materials data facility [63], interatomic potential (force field) databases [64], the Superconducting Material Database (SuperCon) [65], the structural materials database (The Materials Commons) [66], and many others.
2.2. Future trends in materials databases
Currently, there are already a number of materials databases. However, due to non-standardization and fragmentation of material data, many materials databases still cannot satisfy professional needs. In the future, the development trend of materials databases should consider the following aspects:
(1) Diverse storage forms: Databases should be able to store structured, unstructured, and semi-structured data.
(2) Specialized storage categories: Taking research specific materials as the object, a professional database can be established that is highly targeted and easy to use for industry personnel.
(3) Data mining: Using a large amount of stored data in the material database, machine learning can be carried out to discover the relationships among materials composition, process, and performance.
2.3. Material data analytics tools
The interoperability of material datasets and data analytics tools is a critical element of implementing MGI, and material data analytics tools are critical to predicting material performance or designing new materials. Some material data analysis tools are only used internally in the laboratory for reasons of confidentiality, but other data analysis tools are available for purchase.
The Air Force Research Laboratory has developed an integrated collaborative environment that connects laboratory equipment through a laboratory intranet to serve the internal needs of large material laboratories [67].
Microstructure-informed cloud computing provides a practical platform for interoperability of material datasets and data analytics tools [68]. An online computation platform for materials data mining can perform data mining on materials [69]. AiiDA can be used to calculate automatic interactions in materials science [70]. MatCloud is a high-throughput computing infrastructure for the comprehensive management of materials simulation, data, and resources [71]. In addition, the VNL software package provides a graphical environment for first-principles simulations [72].
3. Machine learning as a novel tool
Big Data in materials science cannot directly see the structure of these data using standard tools. Therefore, it is necessary to discover the underlying laws of data based on new methods [73]. The new era of Big Data science and analytics offers a rather compelling approach: machine learning to help with discovery, design, and optimization of novel materials. The machine learning method has developed rapidly, and its long-term goal is the continuous learning and proficiency of the algorithm. For example, without human knowledge, AlphaGo Zero defeated AlphaGo based on a reinforcement learning algorithm, which proved the importance of the continuous learning and progress of the algorithm [74].
Machine learning should be viewed as the sum of the organization of the initial datasets, the descriptor creation and algorithm learning steps, and the necessary subsequent steps for targeted new data entry. Ultimately, the expert recommendation system can be improved continuously and adaptively [43]. Material data are made up of datasets, and dataset uncertainties also have a positive impact on machine learning model predictions [75]. Therefore, Bhat et al. [76] established a set of general rules for describing material data that will aid machine learning and decision-making. Material performance can be predicted through machine learning. In other words, the material is first “fingerprinted” (also called generating a descriptor or a feature vector), followed by a mapping between the descriptor and the property of interest, and finally performance prediction [31,43]. No matter what kind of material is studied, the condition of machine learning is the existence of past data. In machine learning algorithms, the input is generally a component or process parameter of the material, and the output is generally the property or properties of interest [77]. Fig. 1 shows the specific process of machine learning [43]. In Fig. 1, N and M are respectively the number of training examples and the number of fingerprint components.
Fig. 1.
The process of specific machine learning in materials science. (a) Schematic view of an example data set; (b) statement of the learning problem; (c) creation of a surrogate prediction model via the fingerprinting and learning steps [43].
As shown in Fig. 2, machine learning algorithms commonly used in materials science can be divided into four categories: probability estimation [20], regression, clustering, and classification [78]. The main machine learning methods for material performance prediction are regression, clustering, and classification algorithms [20]. Examples include kernel ridge regression [79], artificial neural networks (ANN) [[80], [81], [82]], SVM [83], Gaussian process regression [84], feature learning, clustering, matrix factorization, and constraint reasoning [85], Adaboost [86], and decision trees [87]. Machine learning can also be used to discover new materials, for example, using probability estimation algorithms [88] or principal component analysis [2,89].
Fig. 2.
Common machine learning algorithms in materials science.
Recently, machine learning algorithms and statistical inference [90] have often been used in combination. They are mainly used to predict various properties of material systems effectively and accurately [91]. In the future, they may build material knowledge bases through machine learning and statistical inference [92]. In addition, machine learning methods can be combined with various intelligent algorithms, such as genetic algorithms (GA) [77], which are mainly used to optimize the parameters of machine learning models. Tzuc et al. [93] developed a genetic programming model for optimizing zeolite materials adsorption. Furthermore, GA can explore large and complex search spaces very efficiently [94]. When the amount of data is limited, data mining [95] methods can be used to determine the significant correlations between the ab initio energies of different structures in different materials [2,21,29]. Recently, the method of material data mining has developed rapidly [13] and has produced an open-source toolkit for materials data mining called Matminer [96]. Lu et al. [97] have used data mining to design layered double hydroxides with the required specific surface area.
4. Application fields
There are different types and application fields of machine learning algorithms. The challenge is to judge which machine learning algorithm is the closest to the application field and to maximize prediction accuracy. Then a suitable machine learning model can be created that fits the application field. The application fields of machine learning algorithms in materials science are generally identified as structure prediction, performance prediction, fingerprint prediction, and the new materials discovery, which are introduced one by one below.
4.1. Structure determination
Structural prediction mainly introduces the phase diagram and crystal structure.
4.1.1. Phase diagram determination
High-throughput experimentation studies have resulted in large, rapidly growing volumes of structured data, making expert manual analysis impractical. Therefore, the materials community has turned to fast data analysis techniques for machine learning to convert large amounts of structured data into phase diagrams. Although machine learning promises to provide high-throughput phase diagram determination, its success depends on several factors—prior knowledge, data preprocessing, data representation, similarity or dissimilarity measures, and model choice [85]. Rajan et al. [98] selected principal component analysis, partial least-squares regression, and correlation function expansion to perform data mining on compound semiconductor property phase diagrams. Artrith et al. [99] used a combination ANN and GA algorithm to accelerate sampling of amorphous and disordered materials and constructed a first-principles phase diagram of an amorphous LiSi alloy. This reference showed that using this combined machine learning model can accelerate first-principles sampling of complex structure spaces, proved that this combined model is more efficient than the individual ANN model, and determined that sampling was successful in determining low-energy amorphous structures. Spellings et al. [100] used machine learning to discover interesting regions of the parameter space in colloid self-assembly and created descriptors, then located interesting regions of complex phase diagrams without a priori information, and finally used knowledge of available structures to generate a phase diagram automatically. Fig. 3 shows the phase diagram generated by the Gaussian mixture model.
Fig. 3.
Icosahedral quasicrystal data set phase diagrams generated by unsupervised Gaussian Mixture Models (GMMs).
(a) Shannon entropy (blue line) of the quasicrystal data set as GMM components are successively merged from 15 clusters to one cluster. Merged cluster counts corresponding to (b-d) are indicated by black points. (b-d) Phase diagrams generated by taking the most common predicted cluster type for each parameter point, indicated by the black points in (a). For each selected cluster count, dark gray regions show a poor preference for any single structure among the samples for those parameters. Each type of system, as identified by the GMM is assigned a different color, but this unsupervised algorithm clusters the distinct structures that it finds rather than labeling a previously identified set of known structures. Phase boundaries generated by manual analysis are included for reference as black lines [100].
4.1.2. Crystal structure prediction
Predicting and characterizing the crystal structure of materials is a key issue in materials research and development [2,101,102]. However, the accuracy of the prediction depends largely on how the crystals are represented [103]. Faber et al. [79] introduced and evaluated a set of feature vector representations of crystal structures for machine learning models of formation energies of solids. Faber et al. [104] developed all possible elpasolite crystals using a machine learning model. Data mining was used to establish the structural design rules of crystal chemistry. Combined with first-principles calculations, statistical inference can be used as a tool to accelerate significantly the prediction of unknown crystal structures [105]. Machine learning models can quickly predict the properties of crystalline materials [8]. Furthermore, a machine learning model can also predict point defect properties of crystal structures, which was the first application of machine learning in this field [106].
4.2. Performance prediction
Finding new materials with targeted properties has traditionally been guided by intuition plus trial and error. At present, with the increasing amount of data available, using machine learning to accelerate the prediction of material properties and then to discover these new materials has become a new method. Research on material properties has focused on the relationship between the properties of materials and their microstructure [107,108]. Shi et al. [20] pointed out that machine learning applications to material property prediction can be divided into two categories: macroscopic performance prediction and microscopic property prediction.
4.2.1. Macroscopic performance prediction
Because the machine learning methods of ANN algorithms combined with related optimization algorithms are outstanding in regression and classification problems, they are widely used in performance prediction [20].
An ANN uses massively parallel distributed processors based on data, which tend to store experiential knowledge and make it available for use [109]. ANN theory learns from previously obtained data, which are called training sets, and then checks the system accuracy using test data [110]. ANN is very powerful in solving nonlinear problems [111]. However, artificial neural networks have the drawback of over-fitting, but Bayesian regularization can solve this problem [112].
ANN has been widely used in many fields of research [113]; this may have been due to ANN attempts to model the functioning of human brains [114]. For example, at present, ANN has become a popular tool for Al-Si alloy performance prediction [[115], [116], [117]]. ANN has been developed to predict the porosity percentage of Al-Si casting alloys and has been used to correlate chemical composition and cooling rate with porosity [118,119]. An ANN has accurately predicted the corrosion resistance of Al-Si-Mg-based metal matrix composites reinforced with SiC particles, with the average square of the Pearson product-moment correlation coefficient (R2), the maximum mean square error, and the minimum root mean squared deviation calculated as 0.9904, 0.00002476, and 0.00157480, respectively. It can be seen that the experimental results are highly consistent with the ANN results [120]. ANN was used to predict the mechanical properties of A356, including yield stress, ultimate tensile strength, maximum force, and elongation percentage; the predictions of the ANN model were found to be in good agreement with experimental data [121,122]. In addition, ANN models have been used to investigate the role of composition and processing parameters in the mechanical properties of microalloyed pipeline steel and to design steel with improved performance with regard to strength, impact toughness, and ductility. Then the models were used as objective functions for multi-objective genetic algorithms to evolve tradeoffs among the conflicting objectives of improved strength, better ductility, and higher impact toughness. Moreover, the Pareto optimal solutions were successfully analyzed to study the role of various parameters in designing pipeline steel with such improved performance [77]. Fig. 4 shows the specific flow chart.
Fig. 4.
Flow chart of the computational methodology adopted for designing pipeline steel [77].
Mannodi-Kanakkithodi et al. [123] used a combination of statistical learning and genetic algorithms to predict polymer dielectrics. Within the dataset, the model can not only determine the corresponding properties of any polymer, but can also actively find the specific polymerization that suits its requirements. However, because the model had only seven of the necessary pieces of building-block information, the information guide in the model had certain limitations.
Other applications exist for macroscopic performance prediction. Seko et al. [124] used four regression algorithms to predict the melting temperatures of single and binary compounds. Jha et al. [75] predicted polymer glass transition temperatures. Mauro et al. [125] predicted elastic moduli and compressive stress of glass.
4.2.2. Microscopic property prediction
The microscopic performance of materials depends on their microscopic properties. The application of machine learning to predicting microscopic properties mainly focuses on lattice constants, atomic simulation, and molecular atomization energy [20].
Pilania et al. [126] used statistical learning methods to quickly and accurately predict the various properties of binary wurtzite superlattices. Toyoura et al. [127] used a machine-learning method called the Gaussian process to efficiently identify low-energy regions that characterize proton conduction in the host lattice.
To explore the potential thermal runaway problem of lithium-ion electrode particle microstructure, Petrich et al. [128] used a classification model to detect cracks in lithium-ion batteries and applied the method to real electrode data to test its effectiveness. Deringer et al. [129] developed a machine learning-based Gaussian approximation potential model for atomic simulation of liquid and amorphous elemental carbon. Botu et al. [130] proposed a machine learning method called AGNI - an adaptive, generalizable, and neighborhood informed methodology, which can perform fast and quantum accurate mechanical atomic-force prediction in the neighborhood environment of atoms.
Lilienfeld et al. [131] introduced a machine learning model based on nuclear charges and atomic positions to quickly and accurately predict the atomization energy of various organic molecules and proved its applicability for prediction of molecular atomization potential energy curves. Hansen et al. [132] used machine learning to predict molecular atomization energies, which can significantly accelerate the calculation of quantum chemical properties while maintaining high prediction accuracy. Rupp [25] used kernel ridge regression to predict the atomization energies of small organic molecules. Hansen et al. [133] used machine learning (the so-called Bag of Bonds model; BoB) to estimate molecular atomization energies and predict the exact electronic properties of molecules, Fig. 5 shows that BoB is a stronger machine learning model with proper regularization and improved accuracy by simply expanding the molecular database. Moreover, Pilania et al. [33] and Isayev et al. [134] predicted the bandgap energy.
Fig. 5.
Mean absolute error (MAE in kcal/mol) for BoB and polynomial models: Training sets from N░=░500 to 7000 data points were sampled identically for the different methods. The polynomial models of degree 10 and 18 exhibit high variances due to the random stratification, which for small N leads to nonrobust fits [133].
4.3. Fingerprint (descriptor) prediction
If people can effectively learn from available knowledge and data, the material discovery process can be significantly accelerated and simplified. Using a one-dimensional chain system family, Pilania et al. [91] proposed a general form that can discover decision rules and establish a mapping between attributes that are easily accessible to the system and their properties. The results show that fingerprints based on either chemical structure (compositional and configurational information) or electronic charge density distribution can be used for ultra-fast, but accurate property predictions. Harnessing such learning paradigms extends recent efforts to explore systematically huge chemical spaces and can significantly accelerate discovery of new application-specific materials. Curtarolo et al. [135] introduced new material fingerprint descriptors that produce a material mapping network: nodes represent compounds, and connections represent similarities. The mapping network can identify compounds with different physical and chemical properties. Recently, the t-student stochastic neighbor embedding algorithm was used for the first time to extract feature spaces from band-structure ab initio calculations. This algorithm can design different spaces and map them to lower dimensions, enabling simultaneous analysis and exploration of previously unknown band structures for thousands of materials. The more information is available in the material database, the more space can be explored [136]. Ramprasad et al. [137] have studied a series of motif-based topological fingerprints that can represent the major classes of crystals and molecules by numbers. By using a learning algorithm, these fingerprints can be mapped to various properties of crystals and molecules, thereby accelerating property prediction capabilities. Their paper proves the process by simultaneously optimizing two properties, the gap between the highest occupied and lowest unoccupied molecular orbitals EHL and the isotropic polarizability α. The results are shown in Fig. 6 and Table 1, which show that molecules with the required values of EHL and α were obtained.
Fig. 6.
(Color online) EHL - α log-log plot of the molecule dataset, shown by green symbols, with the predicted fingerprints shown by red diamonds within the regime of desired properties, i.e.,0.6≤α≤0.7░Å3/atom and EHL≥7.0░eV. In the inset, the predicted and calculated properties of the molecules reconstructed from three predicted fingerprints, i.e., C, D, and E, are shown by solid and open symbols: triangles for C, circles for D, and squares for E. The dashed line sketches the limit α ∼ 1/EHL addressed in the text [137].
Table 1
Table 1Predicted and calculated values of α (in Å3/atom) and EHL (in eV) of the molecules designed from three predicted fingerprints, C, D and E. Data from this table are also shown in the inset of Fig. 6 [137].
An adaptive design strategy, tightly coupled with experiments, can accelerate the discovery process by sequentially identifying the next experiments or calculations to effectively navigate the sophisticated search space. The strategy uses inference and global optimization to perform the tradeoff between exploitation and exploration of the search space. Oliynyk et al. [138] used real experimental data, selected a suitable machine learning model, and predicted the model through experiments. Finally, many kinds of intermetallic compounds were discovered. The most crucial finding was RhCd, the first new binary AB compound to be found with a CsCl-type structure in over 15 years. Xue et al. [139] demonstrated this by finding very low thermal hysteresis (DT) NiTi-based shape memory alloys, with Ti50.0Ni46.7Cu0.8Fe2.3Pd0.2 possessing the smallest DT (1.84░K). Wolverton et al. [101] used a general machine learning framework to predict the properties of various materials and discovered a metallic glass alloy. Mauro et al. [125] developed new damage-resistant glasses. Ren et al. [140] found metallic glasses through iterations of machine learning. Yuan et al. [141] used a combination of machine learning and optimization methods to accelerate the discovery of new Pb-free BaTiO3-based piezoelectrics with large electro strains.
Harnessing Big Data, deep data, and smart data from state-of-the-art imaging will accelerate design and discovery of advanced materials. This information is spatially distributed and often has a complex multidimensional nature; hence, the use of unsupervised statistical learning, decorrelation, clustering, and visualization techniques, generally referred to as Big Data approaches, is the first step toward harnessing these data. The correct use of Big Data, or a large amount of data that can be measured and simulated, can serve as a bridge between theory and functional imaging. It can further be extended to what is called deep data by fusing scientific knowledge of the physics and chemistry of the system to Big Data analysis. Interactive knowledge discovery of big and deep data will be made possible by machine learning technologies implemented interactively at all stages of the scientific discovery process from instrument operation to data analysis [4]. Fig. 7 shows the specific process.
Fig. 7.
Imaging techniques (top panels) allow direct measurements of atomic positions and hence bond lengths and angles, local functionalities including chemical states, dielectric properties, and superconductive gap, visualizing atomic, molecular, and defect dynamics in real time, and offers possibilities to control of matter on the molecular and atomic level. In parallel, theoretical methods (bottom panels) allow detailed studies of atomic and electronic structure and dynamics of matter along with prediction of their properties. However, almost lacking are the pathways to bridge theory and experiment. The new advances in data analytics and scientific inference are capable of treating large volumes of data/information and hence linking theory to experiment via microscopic degrees of freedom (middle panels). For example, efficiently matching imaging information about static structure to theoretical simulations on the same material (1░st column) or similarly, the dynamics of oxygen vacancies in materials to corresponding molecular dynamics simulation (3rd column) [4].
To accelerate the design of new materials, recent studies have screened out many types of promising candidate materials [51], including perovskite compounds [83,142], metal-organic frameworks [143,144], catalysts [145,146], light-emitting molecules [147], and thermoelectric materials [[148], [149], [150], [151]]. A large-scale database based on quantum mechanical calculation of known materials, using machine learning to calculate material screening, is a combination screening process for new materials in unconstrained component space, which can be used for an extensive range of critical material discoveries [87]. In addition, interface structure and energy can be also predicted through virtual screening [152].
5. Conclusions
Shared and repeatable material data is the key to advancing material science; the interoperability of material datasets and data analytics tools is a critical element in implementing MGI. Under the premise of complex material data, MGI’s goal is to predict quickly and accurately the various properties of materials and discover new materials. Machine learning methods have taken an essential step toward this goal. Machine learning acts as a bridge in the middle. It uses data to help people discover, design, and optimize new materials, ultimately constituting a material data analysis tool.
At present, the scale of data that can be applied to machine learning in the field of materials is still very small, making it impossible to generate rigorous predictions of material properties and new material discoveries through machine learning. The predicted results are often only close to the probability values of real data and cannot be truly used for experimental guidance. Even if some experts propose to improve machine learning application methods for small-scale datasets, for example by adding a rough attribute estimation to the feature space to build a machine learning model using a small-scale material dataset, the accuracy of the prediction can be improved [153]. However, even with the gradual development of machine learning and deep learning algorithms, from the application experience of each algorithm model, the scale of the dataset is still the main factor in model prediction accuracy. This problem is reflected not only in the machine learning algorithms, but also in deep learning, which has attracted much attention in recent years. In the last few decades, it has been found that material data are presented relatively intact in a large number of scientific papers, and literature data extraction has made some progress in many specific fields, such as chemistry [154] and biomedicine [[155], [156], [157]]. Some studies using these material data have already been carried out. For example, a paper information-assisted extraction software package has been developed and is available online (https://www.mgedata.cn/) [158]. Xie et al. [159] designed the composition of high-performance copper alloys using machine learning based on data from the literature. Edward et al. [42] obtained material synthesis information from academic publications. Kononova et al. [160] obtained inorganic material synthesis recipes by using text mining of scientific publications. However, data in the literature are rarely used by material scientists, and even if the data are used, the data scale is small because there is no mature method to extract data from the material literature. So far, no relatively complete corpus has been developed in the materials field to support the application of material data. Therefore, in the materials genome initiative, material prediction through machine learning should not only focus on research into the machine learning algorithm itself, but should also extract valuable material data from the materials science literature. This would involve first classifying and pre-processing these literature data, then using machine learning algorithms to predict the performance data of interest, and finally finding the correlations among material composition, process, structure, and performance. These steps will play a key role in discovering new properties of materials.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Nos. 61971208, 61671225 and 51864027), the Yunnan Applied Basic Research Projects (No. 2018FA034), the Yunnan Reserve Talents of Young and Middle-aged Academic and Technical Leaders (Shen Tao, 2018), the Yunnan Young Top Talents of Ten Thousands Plan (Shen Tao, Zhu Yan, Yunren Social Development No. 2018 73) and the Scientific Research Foundation of Kunming University of Science and Technology (No. KKSY201703016).
Harnessing big data, deep data, and smart data from state-of-the-art imaging might accelerate the design and realization of advanced functional materials. Here we discuss new opportunities in materials design enabled by the availability of big data in imaging and data analytics approaches, including their limitations, in material systems of practical interest. We specifically focus on how these tools might help realize new discoveries in a timely manner. Such methodologies are particularly appropriate to explore in light of continued improvements in atomistic imaging, modelling and data analytics methods.
Cathode degradation is a key factor that limits the lifetime of Li-ion batteries. To identify functional coatings that can suppress this degradation, we present a high-throughput density functional theory based framework which consists of reaction models that describe thermodynamic and electrochemical stabilities, and acid-scavenging capabilities of materials. Screening more than 130,000 oxygen-bearing materials, we suggest physical and hydrofluoric-acid barrier coatings such as WO3, LiAl5O8 and ZrP2O7 and hydrofluoric-acid scavengers such as Sc2O3, Li2CaGeO4, LiBO2, Li3NbO4, Mg3(BO3)2 and Li2MgSiO4. Using a design strategy to find the thermodynamically optimal coatings for a cathode, we further present optimal hydrofluoric-acid scavengers such as Li2SrSiO4, Li2CaSiO4 and CaIn2O4 for the layered LiCoO2, and Li2GeO3, Li4NiTeO6 and Li2MnO3 for the spinel LiMn2O4 cathodes. These coating materials have the potential to prolong the cycle-life of Li-ion batteries and surpass the performance of common coatings based on conventional materials such as Al2O3, ZnO, MgO or ZrO2.
Machine learning addresses the question of how to build computers that improve automatically through experience. It is one of today's most rapidly growing technical fields, lying at the intersection of computer science and statistics, and at the core of artificial intelligence and data science. Recent progress in machine learning has been driven both by the development of new learning algorithms and theory and by the ongoing explosion in the availability of online data and low-cost computation. The adoption of data-intensive machine-learning methods can be found throughout science, technology and commerce, leading to more evidence-based decision-making across many walks of life, including health care, manufacturing, education, financial modeling, policing, and marketing.
The ability to make rapid and accurate predictions on bandgaps of double perovskites is of much practical interest for a range of applications. While quantum mechanical computations for high-fidelity bandgaps are enormously computation-time intensive and thus impractical in high throughput studies, informatics-based statistical learning approaches can be a promising alternative. Here we demonstrate a systematic feature-engineering approach and a robust learning framework for efficient and accurate predictions of electronic bandgaps of double perovskites. After evaluating a set of more than 1.2 million features, we identify lowest occupied Kohn-Sham levels and elemental electronegativities of the constituent atomic species as the most crucial and relevant predictors. The developed models are validated and tested using the best practices of data science and further analyzed to rationalize their prediction performance.
A long-standing goal of artificial intelligence is an algorithm that learns, tabula rasa, superhuman proficiency in challenging domains. Recently, AlphaGo became the first program to defeat a world champion in the game of Go. The tree search in AlphaGo evaluated positions and selected moves using deep neural networks. These neural networks were trained by supervised learning from human expert moves, and by reinforcement learning from self-play. Here we introduce an algorithm based solely on reinforcement learning, without human data, guidance or domain knowledge beyond game rules. AlphaGo becomes its own teacher: a neural network is trained to predict AlphaGo's own move selections and also the winner of AlphaGo's games. This neural network improves the strength of the tree search, resulting in higher quality move selection and stronger self-play in the next iteration. Starting tabula rasa, our new program AlphaGo Zero achieved superhuman performance, winning 100-0 against the previously published, champion-defeating AlphaGo.
In the paradigm of materials informatics for accelerated materials discovery, the choice of feature set (i.e. attributes that capture aspects of structure, chemistry and/or bonding) is critical. Ideally, the feature sets should provide a simple physical basis for extracting major structural and chemical trends and furthermore, enable rapid predictions of new material chemistries. Orbital radii calculated from model pseudopotential fits to spectroscopic data are potential candidates to satisfy these conditions. Although these radii (and their linear combinations) have been utilized in the past, their functional forms are largely justified with heuristic arguments. Here we show that machine learning methods naturally uncover the functional forms that mimic most frequently used features in the literature, thereby providing a mathematical basis for feature set construction without a priori assumptions. We apply these principles to study two broad materials classes: (i) wide band gap AB compounds and (ii) rare earth-main group RM intermetallics. The AB compounds serve as a prototypical example to demonstrate our approach, whereas the RM intermetallics show how these concepts can be used to rapidly design new ductile materials. Our predictive models indicate that ScCo, ScIr, and YCd should be ductile, whereas each was previously proposed to be brittle.
F.Faber, A.Lindmaa, O.A.von Lilienfeld, R.Armiento, Int. J. Quantum Chem.115(2015) 1094-1101.
Structure quantification is key to successful mining and extraction of core materials knowledge from both multiscale simulations as well as multiscale experiments. The main challenge stems from the need to transform the inherently high dimensional representations demanded by the rich hierarchical material structure into useful, high value, low dimensional representations. In this paper, we develop and demonstrate the merits of a data-driven approach for addressing this challenge at the atomic scale. The approach presented here is built on prior successes demonstrated for mesoscale representations of material internal structure, and involves three main steps: (i) digital representation of the material structure, (ii) extraction of a comprehensive set of structure measures using the framework of n-point spatial correlations, and (iii) identification of data-driven low dimensional measures using principal component analyses. These novel protocols, applied on an ensemble of structure datasets output from molecular dynamics (MD) simulations, have successfully classified the datasets based on several model input parameters such as the interatomic potential and the temperature used in the MD simulations.
The materials discovery process can be significantly expedited and simplified if we can learn effectively from available knowledge and data. In the present contribution, we show that efficient and accurate prediction of a diverse set of properties of material systems is possible by employing machine (or statistical) learning methods trained on quantum mechanical computations in combination with the notions of chemical similarity. Using a family of one-dimensional chain systems, we present a general formalism that allows us to discover decision rules that establish a mapping between easily accessible attributes of a system and its properties. It is shown that fingerprints based on either chemo-structural (compositional and configurational information) or the electronic charge density distribution can be used to make ultra-fast, yet accurate, property predictions. Harnessing such learning paradigms extends recent efforts to systematically explore and mine vast chemical spaces, and can significantly accelerate the discovery of new application-specific materials.
F.Niu, C.Zhang, C.Ré, J.Shavlik, Int. J. Semant. Web Inf.Syst.8(2012) 42-73.
The atomistic modeling of amorphous materials requires structure sizes and sampling statistics that are challenging to achieve with first-principles methods. Here, we propose a methodology to speed up the sampling of amorphous and disordered materials using a combination of a genetic algorithm and a specialized machine-learning potential based on artificial neural networks (ANNs). We show for the example of the amorphous LiSi alloy that around 1000 first-principles calculations are sufficient for the ANN-potential assisted sampling of low-energy atomic configurations in the entire amorphous LixSi phase space. The obtained phase diagram is validated by comparison with the results from an extensive sampling of LixSi configurations using molecular dynamics simulations and a general ANN potential trained to approximately 45 000 first-principles calculations. This demonstrates the utility of the approach for the first-principles modeling of amorphous materials.
Elpasolite is the predominant quaternary crystal structure (AlNaK_{2}F_{6} prototype) reported in the Inorganic Crystal Structure Database. We develop a machine learning model to calculate density functional theory quality formation energies of all approximately 2x10^{6} pristine ABC_{2}D_{6} elpasolite crystals that can be made up from main-group elements (up to bismuth). Our model's accuracy can be improved systematically, reaching a mean absolute error of 0.1 eV/atom for a training set consisting of 10x10^{3} crystals. Important bonding trends are revealed: fluoride is best suited to fit the coordination of the D site, which lowers the formation energy whereas the opposite is found for carbon. The bonding contribution of the elements A and B is very small on average. Low formation energies result from A and B being late elements from group II, C being a late (group I) element, and D being fluoride. Out of 2x10^{6} crystals, 90 unique structures are predicted to be on the convex hull-among which is NFAl_{2}Ca_{6}, with a peculiar stoichiometry and a negative atomic oxidation state for Al.
In this work, it is shown that for the first time that, using information-entropy-based methods, one can quantitatively explore the relative impact of a wide multidimensional array of electronic and chemical bonding parameters on the structural stability of intermetallic compounds. Using an inorganic AB2 compound database as a template data platform, the evolution of design rules for crystal chemistry based on an information-theoretic partitioning classifier for a high-dimensional manifold of crystal chemistry descriptors is monitored. An application of this data-mining approach to establish chemical and structural design rules for crystal chemistry is demonstrated by showing that, when coupled with first-principles calculations, statistical inference methods can serve as a tool for significantly accelerating the prediction of unknown crystal structures.
Artificial neural network (ANN) is a nonlinear dynamic computational system suitable for simulations which are hard to be described by physical models where, rather than relying on a number of predetermined assumptions, data is used to form the model. In order to predict the mechanical properties of A356 including yield stress, ultimate tensile strength and elongation percentage, a relatively new approach that uses artificial neural network and finite element technique is presented which combines mechanical properties data in the form of experimental and simulated solidification conditions. It was observed that predictions of this study are consistent with experimental measurements for A356 alloy. The results of this research were also used for solidification codes of SUT CAST software. Crown Copyright (C) 2011 Published by Elsevier Inc.
M.O.Shabani, A.Mazahery, Metall. Mater. Trans. A43 (2012) 2158-2165.
The ability to efficiently design new and advanced dielectric polymers is hampered by the lack of sufficient, reliable data on wide polymer chemical spaces, and the difficulty of generating such data given time and computational/experimental constraints. Here, we address the issue of accelerating polymer dielectrics design by extracting learning models from data generated by accurate state-of-the-art first principles computations for polymers occupying an important part of the chemical subspace. The polymers are 'fingerprinted' as simple, easily attainable numerical representations, which are mapped to the properties of interest using a machine learning algorithm to develop an on-demand property prediction model. Further, a genetic algorithm is utilised to optimise polymer constituent blocks in an evolutionary manner, thus directly leading to the design of polymers with given target properties. While this philosophy of learning to make instant predictions and design is demonstrated here for the example of polymer dielectrics, it is equally applicable to other classes of materials as well.
The accurate and reliable prediction of properties of molecules typically requires computationally intensive quantum-chemical calculations. Recently, machine learning techniques applied to ab initio calculations have been proposed as an efficient approach for describing the energies of molecules in their given ground-state structure throughout chemical compound space (Rupp et al. Phys. Rev. Lett. 2012, 108, 058301). In this paper we outline a number of established machine learning techniques and investigate the influence of the molecular representation on the methods performance. The best methods achieve prediction errors of 3 kcal/mol for the atomization energies of a wide variety of molecules. Rationales for this performance improvement are given together with pitfalls and challenges when applying machine learning approaches to the prediction of quantum-mechanical observables.
Simultaneously accurate and efficient prediction of molecular properties throughout chemical compound space is a critical ingredient toward rational compound design in chemical and pharmaceutical industries. Aiming toward this goal, we develop and apply a systematic hierarchy of efficient empirical methods to estimate atomization and total energies of molecules. These methods range from a simple sum over atoms, to addition of bond energies, to pairwise interatomic force fields, reaching to the more sophisticated machine learning approaches that are capable of describing collective interactions between many atoms or bonds. In the case of equilibrium molecular geometries, even simple pairwise force fields demonstrate prediction accuracy comparable to benchmark energies calculated using density functional theory with hybrid exchange-correlation functionals; however, accounting for the collective many-body interactions proves to be essential for approaching the
Although historically materials discovery has been driven by a laborious trial-and-error process, knowledge-driven materials design can now be enabled by the rational combination of Machine Learning methods and materials databases. Here, data from the AFLOW repository for ab initio calculations is combined with Quantitative Materials Structure-Property Relationship models to predict important properties: metal/insulator classification, band gap energy, bulk/shear moduli, Debye temperature and heat capacities. The prediction's accuracy compares well with the quality of the training data for virtually any stoichiometric inorganic crystalline material, reciprocating the available thermomechanical experimental data. The universality of the approach is attributed to the construction of the descriptors: Property-Labelled Materials Fragments. The representations require only minimal structural input allowing straightforward implementations of simple heuristic design rules.
Intermetallic compounds are bestowed by diverse compositions, complex structures, and useful properties for many materials applications. How metallic elements react to form these compounds and what structures they adopt remain challenging questions that defy predictability. Traditional approaches offer some rational strategies to prepare specific classes of intermetallics, such as targeting members within a modular homologous series, manipulating building blocks to assemble new structures, and filling interstitial sites to create stuffed variants. Because these strategies rely on precedent, they cannot foresee surprising results, by definition. Exploratory synthesis, whether through systematic phase diagram investigations or serendipity, is still essential for expanding our knowledge base. Eventually, the relationships may become too complex for the pattern recognition skills to be reliably or practically performed by humans. Complementing these traditional approaches, new machine-learning approaches may be a viable alternative for materials discovery, not only among intermetallics but also more generally to other chemical compounds. In this Account, we survey our own efforts to discover new intermetallic compounds, encompassing gallides, germanides, phosphides, arsenides, and others. We apply various machine-learning methods (such as support vector machine and random forest algorithms) to confront two significant questions in solid state chemistry. First, what crystal structures are adopted by a compound given an arbitrary composition? Initial efforts have focused on binary equiatomic phases AB, ternary equiatomic phases ABC, and full Heusler phases AB2C. Our analysis emphasizes the use of real experimental data and places special value on confirming predictions through experiment. Chemical descriptors are carefully chosen through a rigorous procedure called cluster resolution feature selection. Predictions for crystal structures are quantified by evaluating probabilities. Major results include the discovery of RhCd, the first new binary AB compound to be found in over 15 years, with a CsCl-type structure; the connection between
With more than a hundred elements in the periodic table, a large number of potential new materials exist to address the technological and societal challenges we face today; however, without some guidance, searching through this vast combinatorial space is frustratingly slow and expensive, especially for materials strongly influenced by processing. We train a machine learning (ML) model on previously reported observations, parameters from physiochemical theories, and make it synthesis method-dependent to guide high-throughput (HiTp) experiments to find a new system of metallic glasses in the Co-V-Zr ternary. Experimental observations are in good agreement with the predictions of the model, but there are quantitative discrepancies in the precise compositions predicted. We use these discrepancies to retrain the ML model. The refined model has significantly improved accuracy not only for the Co-V-Zr system but also across all other available validation data. We then use the refined model to guide the discovery of metallic glasses in two additional previously unreported ternaries. Although our approach of iterative use of ML and HiTp experiments has guided us to rapid discovery of three new glass-forming systems, it has also provided us with a quantitatively accurate, synthesis method-sensitive predictor for metallic glasses that improves performance with use and thus promises to greatly accelerate discovery of many new metallic glasses. We believe that this discovery paradigm is applicable to a wider range of materials and should prove equally powerful for other materials and properties that are synthesis path-dependent and that current physiochemical theories find challenging to predict.
One of the main bottlenecks to deploying large-scale carbon dioxide capture and storage (CCS) in power plants is the energy required to separate the CO(2) from flue gas. For example, near-term CCS technology applied to coal-fired power plants is projected to reduce the net output of the plant by some 30% and to increase the cost of electricity by 60-80%. Developing capture materials and processes that reduce the parasitic energy imposed by CCS is therefore an important area of research. We have developed a computational approach to rank adsorbents for their performance in CCS. Using this analysis, we have screened hundreds of thousands of zeolite and zeolitic imidazolate framework structures and identified many different structures that have the potential to reduce the parasitic energy of CCS by 30-40% compared with near-term technologies.
Metal-organic frameworks (MOFs) are porous materials constructed from modular molecular building blocks, typically metal clusters and organic linkers. These can, in principle, be assembled to form an almost unlimited number of MOFs, yet materials reported to date represent only a tiny fraction of the possible combinations. Here, we demonstrate a computational approach to generate all conceivable MOFs from a given chemical library of building blocks (based on the structures of known MOFs) and rapidly screen them to find the best candidates for a specific application. From a library of 102 building blocks we generated 137,953 hypothetical MOFs and for each one calculated the pore-size distribution, surface area and methane-storage capacity. We identified over 300 MOFs with a predicted methane-storage capacity better than that of any known material, and this approach also revealed structure-property relationships. Methyl-functionalized MOFs were frequently top performers, so we selected one such promising MOF and experimentally confirmed its predicted capacity.
The pace of materials discovery for heterogeneous catalysts and electrocatalysts could, in principle, be accelerated by the development of efficient computational screening methods. This would require an integrated approach, where the catalytic activity and stability of new materials are evaluated and where predictions are benchmarked by careful synthesis and experimental tests. In this contribution, we present a density functional theory-based, high-throughput screening scheme that successfully uses these strategies to identify a new electrocatalyst for the hydrogen evolution reaction (HER). The activity of over 700 binary surface alloys is evaluated theoretically; the stability of each alloy in electrochemical environments is also estimated. BiPt is found to have a predicted activity comparable to, or even better than, pure Pt, the archetypical HER catalyst. This alloy is synthesized and tested experimentally and shows improved HER performance compared with pure Pt, in agreement with the computational screening results.
W.T.Hong, R.E.Welsch, S.-H.Yang, J. Phys. Chem. C120 (2016) 78-86.
Virtual screening is becoming a ground-breaking tool for molecular discovery due to the exponential growth of available computer time and constant improvement of simulation and machine learning techniques. We report an integrated organic functional material design process that incorporates theoretical insight, quantum chemistry, cheminformatics, machine learning, industrial expertise, organic synthesis, molecular characterization, device fabrication and optoelectronic testing. After exploring a search space of 1.6 million molecules and screening over 400,000 of them using time-dependent density functional theory, we identified thousands of promising novel organic light-emitting diode molecules across the visible spectrum. Our team collaboratively selected the best candidates from this set. The experimentally determined external quantum efficiencies for these synthesized candidates were as large as 22%.
Interfaces markedly affect the properties of materials because of differences in their atomic configurations. Determining the atomic structure of the interface is therefore one of the most significant tasks in materials research. However, determining the interface structure usually requires extensive computation. If the interface structure could be efficiently predicted, our understanding of the mechanisms that give rise to the interface properties would be significantly facilitated, and this would pave the way for the design of material interfaces. Using a virtual screening method based on machine learning, we demonstrate a powerful technique to determine interface energies and structures. On the basis of the results obtained by a nonlinear regression using training data from 4 interfaces, structures and energies for 13 other interfaces were predicted. Our method achieved an efficiency that is more than several hundred to several tens of thousand times higher than that of the previously reported methods. Because the present method uses geometrical factors, such as bond length and atomic density, as descriptors for the regression analysis, the method presented here is robust and general and is expected to be beneficial to understanding the nature of any interface.
Efficient access to chemical information contained in scientific literature, patents, technical reports, or the web is a pressing need shared by researchers and patent attorneys from different chemical disciplines. Retrieval of important chemical information in most cases starts with finding relevant documents for a particular chemical compound or family. Targeted retrieval of chemical documents is closely connected to the automatic recognition of chemical entities in the text, which commonly involves the extraction of the entire list of chemicals mentioned in a document, including any associated information. In this Review, we provide a comprehensive and in-depth description of fundamental concepts, technical implementations, and current technologies for meeting these information demands. A strong focus is placed on community challenges addressing systems performance, more particularly CHEMDNER and CHEMDNER patents tasks of BioCreative IV and V, respectively. Considering the growing interest in the construction of automatically annotated chemical knowledge bases that integrate chemical information and biological data, cheminformatics approaches for mapping the extracted chemical names into chemical structures and their subsequent annotation together with text mining applications for linking chemistry with biological information are also presented. Finally, future trends and current challenges are highlighted as a roadmap proposal for research in this emerging field.
Manually curating knowledge from biomedical literature into structured databases is highly expensive and time-consuming, making it difficult to keep pace with the rapid growth of the literature. There is therefore a pressing need to assist biocuration with automated text mining tools. Here, we describe PubTator, a web-based system for assisting biocuration. PubTator is different from the few existing tools by featuring a PubMed-like interface, which many biocurators find familiar, and being equipped with multiple challenge-winning text mining algorithms to ensure the quality of its automatic results. Through a formal evaluation with two external user groups, PubTator was shown to be capable of improving both the efficiency and accuracy of manual curation. PubTator is publicly available at http://www.ncbi.nlm.nih.gov/CBBresearch/Lu/Demo/PubTator/.
TRIM-NHL proteins are conserved regulators of development and differentiation but their molecular function has remained largely elusive. Here, we report an as yet unrecognized activity for the mammalian TRIM-NHL protein TRIM71 as a repressor of mRNAs. We show that TRIM71 is associated with mRNAs and that it promotes translational repression and mRNA decay. We have identified Rbl1 and Rbl2, two transcription factors whose down-regulation is important for stem cell function, as TRIM71 targets in mouse embryonic stem cells. Furthermore, one of the defining features of TRIM-NHL proteins, the NHL domain, is necessary and sufficient to target TRIM71 to RNA, while the RING domain that confers ubiquitin ligase activity is dispensable for repression. Our results reveal strong similarities between TRIM71 and Drosophila BRAT, the best-studied TRIM-NHL protein and a well-documented translational repressor, suggesting that BRAT and TRIM71 are part of a family of mRNA repressors regulating proliferation and differentiation.
Paper Information Assisted Extraction Software, 2019, November 12 https://www.mgedata.cn.
Materials discovery has become significantly facilitated and accelerated by high-throughput ab-initio computations. This ability to rapidly design interesting novel compounds has displaced the materials innovation bottleneck to the development of synthesis routes for the desired material. As there is no a fundamental theory for materials synthesis, one might attempt a data-driven approach for predicting inorganic materials synthesis, but this is impeded by the lack of a comprehensive database containing synthesis processes. To overcome this limitation, we have generated a dataset of
5
2016
... At the beginning of the 21st Century, Ceder, who worked at the Massachusetts Institute of Technology (MIT; Cambridge, MA, USA) was influenced by high-throughput, data-driven material discovery methods and inspired by the Human Genome Project. He considered whether material scientists could learn from the experience of geneticists. In 2006, Ceder launched the Materials Genomics project at the MIT, using an improved data-mining algorithm to predict lithium-based materials for electric vehicle batteries. By 2010, the project had evolved to include approximately 20,000 predicted compounds [1]. At the same time, at Duke University (Durham, NC, USA), Curtarolo established the Materials Genomics Center, focusing on metal alloy research. His team gradually extended the 2003 algorithm and library [2] to AFLOW (Automatic Flow), a system that can calculate known crystal structures and automatically predict new structures [3]. Despite this, computational materials science was still not valued by experts until the American White House announced hundreds of millions of dollars of funding for the Materials Genome Initiative (MGI) in June 2011 [1]. ...
... ]. Despite this, computational materials science was still not valued by experts until the American White House announced hundreds of millions of dollars of funding for the Materials Genome Initiative (MGI) in June 2011 [1]. ...
... Continuous advancement in science depends on shared and repeatable data [36]. Large-scale data analysis is the driving force for material progress. Materials science is on the verge of adopting Materials 4.0 /fourth paradigm [37,38] or data-driven [39] discovery on large-scale data [40]. The quality of data analysis and data mining is affected by the data quality of the materials database and directly affects the application of data that are mined. The existing database is far from including all known materials, not to mention all possible materials. Data-driven discovery applies to certain materials, but not to all materials. Even if people pick an interesting material on their computers, it will take years to synthesize it in the laboratory [1]. ...
... An early database, The Inorganic Crystal Structure Database [45], created in 2002, which covers the literature from 1915 to 2002, is a comprehensive collection of crystal structure information for non-organic compounds, including inorganics, minerals, ceramics, and metals [46]. In 2015, Wolverton [47] launched the Open Quantum Materials Database (OQMD), which is also an inorganic crystal structure database with approximately 50,000 pieces of data from a widely used experimental library [1]. Cambridge Structural Databases (CSD) [48] is a worldwide repository for small-molecule organic and metal-organic crystal structures hosted by Cambridge Crystallographic Data Center (CCDC) (https://www.lib.ncsu.edu/databases/cambridge-structural.html [49]). The Materials Projec t [50,51] is the core program of the Materials Genome Initiative, which uses high-throughput calculations to reveal the properties of all known inorganic materials. These open datasets can be accessed through multiple channels for interactive exploration and data mining. The Materials Project URL is https://www.materialsproject.org [52]. By September 2019, the database contained 120,612 inorganic compounds, 52,366 band structures, 35,336 molecules, 530,243 nanoporous materials, 13,621 elastic tensors, 3003 piezoelectric tensors, 4401 intercalation electrodes, and 16,128 conversion electrodes. ...
... In 2003, Ceder et al. [1] first demonstrated how quantum mechanical computing databases could help predict the most likely crystal structure of metal alloys—a crucial step in the invention of new materials. Ceder introduced the concept of public libraries of material properties and used data mining to fill in missing data. In the second year after the genome project was launched, Curtarolo [53] posted a database based on his software, called AFLOWlib, which is a database with more than one million materials and approximately 100 million calculated properties. AFLOWlib contains such a large amount of data because it also includes thousands of hypothetical materials, many of which exist for only one second in the real world, but Curtarolo used these data to study why some alloys can form metallic glass [54]. OQMD is also a high-throughput database that includes a large number of hypothetical materials, and the entire database can be obtained for free at http://www.oqmd.org/download [47]. ...
4
2003
... At the beginning of the 21st Century, Ceder, who worked at the Massachusetts Institute of Technology (MIT; Cambridge, MA, USA) was influenced by high-throughput, data-driven material discovery methods and inspired by the Human Genome Project. He considered whether material scientists could learn from the experience of geneticists. In 2006, Ceder launched the Materials Genomics project at the MIT, using an improved data-mining algorithm to predict lithium-based materials for electric vehicle batteries. By 2010, the project had evolved to include approximately 20,000 predicted compounds [1]. At the same time, at Duke University (Durham, NC, USA), Curtarolo established the Materials Genomics Center, focusing on metal alloy research. His team gradually extended the 2003 algorithm and library [2] to AFLOW (Automatic Flow), a system that can calculate known crystal structures and automatically predict new structures [3]. Despite this, computational materials science was still not valued by experts until the American White House announced hundreds of millions of dollars of funding for the Materials Genome Initiative (MGI) in June 2011 [1]. ...
... As shown in Fig. 2, machine learning algorithms commonly used in materials science can be divided into four categories: probability estimation [20], regression, clustering, and classification [78]. The main machine learning methods for material performance prediction are regression, clustering, and classification algorithms [20]. Examples include kernel ridge regression [79], artificial neural networks (ANN) [[80], [81], [82]], SVM [83], Gaussian process regression [84], feature learning, clustering, matrix factorization, and constraint reasoning [85], Adaboost [86], and decision trees [87]. Machine learning can also be used to discover new materials, for example, using probability estimation algorithms [88] or principal component analysis [2,89]. ...
... Recently, machine learning algorithms and statistical inference [90] have often been used in combination. They are mainly used to predict various properties of material systems effectively and accurately [91]. In the future, they may build material knowledge bases through machine learning and statistical inference [92]. In addition, machine learning methods can be combined with various intelligent algorithms, such as genetic algorithms (GA) [77], which are mainly used to optimize the parameters of machine learning models. Tzuc et al. [93] developed a genetic programming model for optimizing zeolite materials adsorption. Furthermore, GA can explore large and complex search spaces very efficiently [94]. When the amount of data is limited, data mining [95] methods can be used to determine the significant correlations between the ab initio energies of different structures in different materials [2,21,29]. Recently, the method of material data mining has developed rapidly [13] and has produced an open-source toolkit for materials data mining called Matminer [96]. Lu et al. [97] have used data mining to design layered double hydroxides with the required specific surface area. ...
... Predicting and characterizing the crystal structure of materials is a key issue in materials research and development [2,101,102]. However, the accuracy of the prediction depends largely on how the crystals are represented [103]. Faber et al. [79] introduced and evaluated a set of feature vector representations of crystal structures for machine learning models of formation energies of solids. Faber et al. [104] developed all possible elpasolite crystals using a machine learning model. Data mining was used to establish the structural design rules of crystal chemistry. Combined with first-principles calculations, statistical inference can be used as a tool to accelerate significantly the prediction of unknown crystal structures [105]. Machine learning models can quickly predict the properties of crystalline materials [8]. Furthermore, a machine learning model can also predict point defect properties of crystal structures, which was the first application of machine learning in this field [106]. ...
1
2012
... At the beginning of the 21st Century, Ceder, who worked at the Massachusetts Institute of Technology (MIT; Cambridge, MA, USA) was influenced by high-throughput, data-driven material discovery methods and inspired by the Human Genome Project. He considered whether material scientists could learn from the experience of geneticists. In 2006, Ceder launched the Materials Genomics project at the MIT, using an improved data-mining algorithm to predict lithium-based materials for electric vehicle batteries. By 2010, the project had evolved to include approximately 20,000 predicted compounds [1]. At the same time, at Duke University (Durham, NC, USA), Curtarolo established the Materials Genomics Center, focusing on metal alloy research. His team gradually extended the 2003 algorithm and library [2] to AFLOW (Automatic Flow), a system that can calculate known crystal structures and automatically predict new structures [3]. Despite this, computational materials science was still not valued by experts until the American White House announced hundreds of millions of dollars of funding for the Materials Genome Initiative (MGI) in June 2011 [1]. ...
3
2015
... As highlighted by the MGI, theory and simulation can enhance the efficiency of materials discovery and design. On the other hand, a purely theoretical and simulation approach to materials discovery and design is often tricky to realize experimentally due to complications in synthesis, existence, and production of defects and possible metastable states. To bridge this gap, all aspects of theory and simulation must be tightly integrated with experiments and data [4]. High-throughput density functional theory has become a universal tool for material discovery [[5], [6], [7]], but its limitation is that computational costs are high [8]. The need now is to overcome this limitation to take advantage of the latest advances in data and information science [9]. ...
... Harnessing Big Data, deep data, and smart data from state-of-the-art imaging will accelerate design and discovery of advanced materials. This information is spatially distributed and often has a complex multidimensional nature; hence, the use of unsupervised statistical learning, decorrelation, clustering, and visualization techniques, generally referred to as Big Data approaches, is the first step toward harnessing these data. The correct use of Big Data, or a large amount of data that can be measured and simulated, can serve as a bridge between theory and functional imaging. It can further be extended to what is called deep data by fusing scientific knowledge of the physics and chemistry of the system to Big Data analysis. Interactive knowledge discovery of big and deep data will be made possible by machine learning technologies implemented interactively at all stages of the scientific discovery process from instrument operation to data analysis [4]. Fig. 7 shows the specific process. ...
... Imaging techniques (top panels) allow direct measurements of atomic positions and hence bond lengths and angles, local functionalities including chemical states, dielectric properties, and superconductive gap, visualizing atomic, molecular, and defect dynamics in real time, and offers possibilities to control of matter on the molecular and atomic level. In parallel, theoretical methods (bottom panels) allow detailed studies of atomic and electronic structure and dynamics of matter along with prediction of their properties. However, almost lacking are the pathways to bridge theory and experiment. The new advances in data analytics and scientific inference are capable of treating large volumes of data/information and hence linking theory to experiment via microscopic degrees of freedom (middle panels). For example, efficiently matching imaging information about static structure to theoretical simulations on the same material (1░st column) or similarly, the dynamics of oxygen vacancies in materials to corresponding molecular dynamics simulation (3rd column) [4].
To accelerate the design of new materials, recent studies have screened out many types of promising candidate materials [51], including perovskite compounds [83,142], metal-organic frameworks [143,144], catalysts [145,146], light-emitting molecules [147], and thermoelectric materials [[148], [149], [150], [151]]. A large-scale database based on quantum mechanical calculation of known materials, using machine learning to calculate material screening, is a combination screening process for new materials in unconstrained component space, which can be used for an extensive range of critical material discoveries [87]. In addition, interface structure and energy can be also predicted through virtual screening [152]. ...
1
2016
... As highlighted by the MGI, theory and simulation can enhance the efficiency of materials discovery and design. On the other hand, a purely theoretical and simulation approach to materials discovery and design is often tricky to realize experimentally due to complications in synthesis, existence, and production of defects and possible metastable states. To bridge this gap, all aspects of theory and simulation must be tightly integrated with experiments and data [4]. High-throughput density functional theory has become a universal tool for material discovery [[5], [6], [7]], but its limitation is that computational costs are high [8]. The need now is to overcome this limitation to take advantage of the latest advances in data and information science [9]. ...
1
2017
... As highlighted by the MGI, theory and simulation can enhance the efficiency of materials discovery and design. On the other hand, a purely theoretical and simulation approach to materials discovery and design is often tricky to realize experimentally due to complications in synthesis, existence, and production of defects and possible metastable states. To bridge this gap, all aspects of theory and simulation must be tightly integrated with experiments and data [4]. High-throughput density functional theory has become a universal tool for material discovery [[5], [6], [7]], but its limitation is that computational costs are high [8]. The need now is to overcome this limitation to take advantage of the latest advances in data and information science [9]. ...
1
2016
... As highlighted by the MGI, theory and simulation can enhance the efficiency of materials discovery and design. On the other hand, a purely theoretical and simulation approach to materials discovery and design is often tricky to realize experimentally due to complications in synthesis, existence, and production of defects and possible metastable states. To bridge this gap, all aspects of theory and simulation must be tightly integrated with experiments and data [4]. High-throughput density functional theory has become a universal tool for material discovery [[5], [6], [7]], but its limitation is that computational costs are high [8]. The need now is to overcome this limitation to take advantage of the latest advances in data and information science [9]. ...
2
2017
... As highlighted by the MGI, theory and simulation can enhance the efficiency of materials discovery and design. On the other hand, a purely theoretical and simulation approach to materials discovery and design is often tricky to realize experimentally due to complications in synthesis, existence, and production of defects and possible metastable states. To bridge this gap, all aspects of theory and simulation must be tightly integrated with experiments and data [4]. High-throughput density functional theory has become a universal tool for material discovery [[5], [6], [7]], but its limitation is that computational costs are high [8]. The need now is to overcome this limitation to take advantage of the latest advances in data and information science [9]. ...
... Predicting and characterizing the crystal structure of materials is a key issue in materials research and development [2,101,102]. However, the accuracy of the prediction depends largely on how the crystals are represented [103]. Faber et al. [79] introduced and evaluated a set of feature vector representations of crystal structures for machine learning models of formation energies of solids. Faber et al. [104] developed all possible elpasolite crystals using a machine learning model. Data mining was used to establish the structural design rules of crystal chemistry. Combined with first-principles calculations, statistical inference can be used as a tool to accelerate significantly the prediction of unknown crystal structures [105]. Machine learning models can quickly predict the properties of crystalline materials [8]. Furthermore, a machine learning model can also predict point defect properties of crystal structures, which was the first application of machine learning in this field [106]. ...
1
2016
... As highlighted by the MGI, theory and simulation can enhance the efficiency of materials discovery and design. On the other hand, a purely theoretical and simulation approach to materials discovery and design is often tricky to realize experimentally due to complications in synthesis, existence, and production of defects and possible metastable states. To bridge this gap, all aspects of theory and simulation must be tightly integrated with experiments and data [4]. High-throughput density functional theory has become a universal tool for material discovery [[5], [6], [7]], but its limitation is that computational costs are high [8]. The need now is to overcome this limitation to take advantage of the latest advances in data and information science [9]. ...
1
2016
... Machine learning [10] is an interdisciplinary subject lying between computer science and statistics. It is the core of artificial intelligence and data science. It promotes the availability of online data and low-cost computing through algorithmic learning [11]. Even though machine learning is a rapidly innovative technology, some algorithms in machine learning are bound to be outdated, but the importance of using scientific reasoning to find reliable structure, property, and processing models will never be outdated [12]. The types of materials are diverse, and the factors affecting the results are complex. Therefore, the data relationship needs to be clear, and the machine learning method is good at discovering and establishing connections among numerous data points. The introduction of machine learning methods is beneficial in understanding and discovering the correlation between experimental parameters and materials performance. ...
1
2015
... Machine learning [10] is an interdisciplinary subject lying between computer science and statistics. It is the core of artificial intelligence and data science. It promotes the availability of online data and low-cost computing through algorithmic learning [11]. Even though machine learning is a rapidly innovative technology, some algorithms in machine learning are bound to be outdated, but the importance of using scientific reasoning to find reliable structure, property, and processing models will never be outdated [12]. The types of materials are diverse, and the factors affecting the results are complex. Therefore, the data relationship needs to be clear, and the machine learning method is good at discovering and establishing connections among numerous data points. The introduction of machine learning methods is beneficial in understanding and discovering the correlation between experimental parameters and materials performance. ...
1
2016
... Machine learning [10] is an interdisciplinary subject lying between computer science and statistics. It is the core of artificial intelligence and data science. It promotes the availability of online data and low-cost computing through algorithmic learning [11]. Even though machine learning is a rapidly innovative technology, some algorithms in machine learning are bound to be outdated, but the importance of using scientific reasoning to find reliable structure, property, and processing models will never be outdated [12]. The types of materials are diverse, and the factors affecting the results are complex. Therefore, the data relationship needs to be clear, and the machine learning method is good at discovering and establishing connections among numerous data points. The introduction of machine learning methods is beneficial in understanding and discovering the correlation between experimental parameters and materials performance. ...
2
2017
... The main processes of machine learning may be divided into data preparation, descriptor selection, algorithm/model selection, model prediction, and model application [13]. By applying this process to material discovery and design, a complete cycle is fulfilled, from experimental process data collection to performance prediction and finally to experimental validation. Machine learning methods provide an efficient toolset for extracting important correlations among material phenomena [14]. Machine learning can classify viral genomes [15] and identify type 2 diabetes [16]. In the field of chemoinformatics, machine learning models can detect outlier samples [17] and analyze chemical data [18]. It can even perform classification of potato bruise levels [19]. It is evident that machine learning methods have attracted the attention of researchers in many fields. The approach has been widely used in biology, medicine, and other research fields. In the field of materials science, some materials researchers have used machine learning methods to predict material properties. ...
... Recently, machine learning algorithms and statistical inference [90] have often been used in combination. They are mainly used to predict various properties of material systems effectively and accurately [91]. In the future, they may build material knowledge bases through machine learning and statistical inference [92]. In addition, machine learning methods can be combined with various intelligent algorithms, such as genetic algorithms (GA) [77], which are mainly used to optimize the parameters of machine learning models. Tzuc et al. [93] developed a genetic programming model for optimizing zeolite materials adsorption. Furthermore, GA can explore large and complex search spaces very efficiently [94]. When the amount of data is limited, data mining [95] methods can be used to determine the significant correlations between the ab initio energies of different structures in different materials [2,21,29]. Recently, the method of material data mining has developed rapidly [13] and has produced an open-source toolkit for materials data mining called Matminer [96]. Lu et al. [97] have used data mining to design layered double hydroxides with the required specific surface area. ...
1
2018
... The main processes of machine learning may be divided into data preparation, descriptor selection, algorithm/model selection, model prediction, and model application [13]. By applying this process to material discovery and design, a complete cycle is fulfilled, from experimental process data collection to performance prediction and finally to experimental validation. Machine learning methods provide an efficient toolset for extracting important correlations among material phenomena [14]. Machine learning can classify viral genomes [15] and identify type 2 diabetes [16]. In the field of chemoinformatics, machine learning models can detect outlier samples [17] and analyze chemical data [18]. It can even perform classification of potato bruise levels [19]. It is evident that machine learning methods have attracted the attention of researchers in many fields. The approach has been widely used in biology, medicine, and other research fields. In the field of materials science, some materials researchers have used machine learning methods to predict material properties. ...
1
2017
... The main processes of machine learning may be divided into data preparation, descriptor selection, algorithm/model selection, model prediction, and model application [13]. By applying this process to material discovery and design, a complete cycle is fulfilled, from experimental process data collection to performance prediction and finally to experimental validation. Machine learning methods provide an efficient toolset for extracting important correlations among material phenomena [14]. Machine learning can classify viral genomes [15] and identify type 2 diabetes [16]. In the field of chemoinformatics, machine learning models can detect outlier samples [17] and analyze chemical data [18]. It can even perform classification of potato bruise levels [19]. It is evident that machine learning methods have attracted the attention of researchers in many fields. The approach has been widely used in biology, medicine, and other research fields. In the field of materials science, some materials researchers have used machine learning methods to predict material properties. ...
1
2017
... The main processes of machine learning may be divided into data preparation, descriptor selection, algorithm/model selection, model prediction, and model application [13]. By applying this process to material discovery and design, a complete cycle is fulfilled, from experimental process data collection to performance prediction and finally to experimental validation. Machine learning methods provide an efficient toolset for extracting important correlations among material phenomena [14]. Machine learning can classify viral genomes [15] and identify type 2 diabetes [16]. In the field of chemoinformatics, machine learning models can detect outlier samples [17] and analyze chemical data [18]. It can even perform classification of potato bruise levels [19]. It is evident that machine learning methods have attracted the attention of researchers in many fields. The approach has been widely used in biology, medicine, and other research fields. In the field of materials science, some materials researchers have used machine learning methods to predict material properties. ...
1
2018
... The main processes of machine learning may be divided into data preparation, descriptor selection, algorithm/model selection, model prediction, and model application [13]. By applying this process to material discovery and design, a complete cycle is fulfilled, from experimental process data collection to performance prediction and finally to experimental validation. Machine learning methods provide an efficient toolset for extracting important correlations among material phenomena [14]. Machine learning can classify viral genomes [15] and identify type 2 diabetes [16]. In the field of chemoinformatics, machine learning models can detect outlier samples [17] and analyze chemical data [18]. It can even perform classification of potato bruise levels [19]. It is evident that machine learning methods have attracted the attention of researchers in many fields. The approach has been widely used in biology, medicine, and other research fields. In the field of materials science, some materials researchers have used machine learning methods to predict material properties. ...
1
2018
... The main processes of machine learning may be divided into data preparation, descriptor selection, algorithm/model selection, model prediction, and model application [13]. By applying this process to material discovery and design, a complete cycle is fulfilled, from experimental process data collection to performance prediction and finally to experimental validation. Machine learning methods provide an efficient toolset for extracting important correlations among material phenomena [14]. Machine learning can classify viral genomes [15] and identify type 2 diabetes [16]. In the field of chemoinformatics, machine learning models can detect outlier samples [17] and analyze chemical data [18]. It can even perform classification of potato bruise levels [19]. It is evident that machine learning methods have attracted the attention of researchers in many fields. The approach has been widely used in biology, medicine, and other research fields. In the field of materials science, some materials researchers have used machine learning methods to predict material properties. ...
1
2018
... The main processes of machine learning may be divided into data preparation, descriptor selection, algorithm/model selection, model prediction, and model application [13]. By applying this process to material discovery and design, a complete cycle is fulfilled, from experimental process data collection to performance prediction and finally to experimental validation. Machine learning methods provide an efficient toolset for extracting important correlations among material phenomena [14]. Machine learning can classify viral genomes [15] and identify type 2 diabetes [16]. In the field of chemoinformatics, machine learning models can detect outlier samples [17] and analyze chemical data [18]. It can even perform classification of potato bruise levels [19]. It is evident that machine learning methods have attracted the attention of researchers in many fields. The approach has been widely used in biology, medicine, and other research fields. In the field of materials science, some materials researchers have used machine learning methods to predict material properties. ...
6
2017
... Recently, material discovery and design using machine learning have received increasing attention and have been greatly improved from the standpoints of time efficiency and prediction accuracy [20,21]. Voyles [22] used machine learning algorithms to improve the quality of material microscope data and extract material information from enhanced data. Raccuglia et al. [23] used the support vector machine (SVM) approach to predict the feasibility of untested responses to data collected in failed experiments. Wicker et al. [24] used the SVM algorithm with an RBF kernel to predict the crystallinity of molecular materials. Rupp [25] found that combining quantum mechanics (QM) with machine learning could be expected to improve the accuracy of QM. Stanev et al. [26] proposed that a machine learning model could simulate the critical temperature of superconductors. A neural network can represent high-dimensional potential energy surfaces when the atomistic materials are simulated, and machine-learned interpolation of atomic potential energy surfaces can automatically construct high-precision atomic interaction potentials [[27], [28], [29]]. The predictive power of machine learning algorithms was also reflected in many aspects of material properties, such as sintered density [30], atomization energies [31], band gaps [32,33], and phase transitions [34,35]. ...
... As shown in Fig. 2, machine learning algorithms commonly used in materials science can be divided into four categories: probability estimation [20], regression, clustering, and classification [78]. The main machine learning methods for material performance prediction are regression, clustering, and classification algorithms [20]. Examples include kernel ridge regression [79], artificial neural networks (ANN) [[80], [81], [82]], SVM [83], Gaussian process regression [84], feature learning, clustering, matrix factorization, and constraint reasoning [85], Adaboost [86], and decision trees [87]. Machine learning can also be used to discover new materials, for example, using probability estimation algorithms [88] or principal component analysis [2,89]. ...
... ]. The main machine learning methods for material performance prediction are regression, clustering, and classification algorithms [20]. Examples include kernel ridge regression [79], artificial neural networks (ANN) [[80], [81], [82]], SVM [83], Gaussian process regression [84], feature learning, clustering, matrix factorization, and constraint reasoning [85], Adaboost [86], and decision trees [87]. Machine learning can also be used to discover new materials, for example, using probability estimation algorithms [88] or principal component analysis [2,89]. ...
... Finding new materials with targeted properties has traditionally been guided by intuition plus trial and error. At present, with the increasing amount of data available, using machine learning to accelerate the prediction of material properties and then to discover these new materials has become a new method. Research on material properties has focused on the relationship between the properties of materials and their microstructure [107,108]. Shi et al. [20] pointed out that machine learning applications to material property prediction can be divided into two categories: macroscopic performance prediction and microscopic property prediction. ...
... Because the machine learning methods of ANN algorithms combined with related optimization algorithms are outstanding in regression and classification problems, they are widely used in performance prediction [20]. ...
... The microscopic performance of materials depends on their microscopic properties. The application of machine learning to predicting microscopic properties mainly focuses on lattice constants, atomic simulation, and molecular atomization energy [20]. ...
2
2017
... Recently, material discovery and design using machine learning have received increasing attention and have been greatly improved from the standpoints of time efficiency and prediction accuracy [20,21]. Voyles [22] used machine learning algorithms to improve the quality of material microscope data and extract material information from enhanced data. Raccuglia et al. [23] used the support vector machine (SVM) approach to predict the feasibility of untested responses to data collected in failed experiments. Wicker et al. [24] used the SVM algorithm with an RBF kernel to predict the crystallinity of molecular materials. Rupp [25] found that combining quantum mechanics (QM) with machine learning could be expected to improve the accuracy of QM. Stanev et al. [26] proposed that a machine learning model could simulate the critical temperature of superconductors. A neural network can represent high-dimensional potential energy surfaces when the atomistic materials are simulated, and machine-learned interpolation of atomic potential energy surfaces can automatically construct high-precision atomic interaction potentials [[27], [28], [29]]. The predictive power of machine learning algorithms was also reflected in many aspects of material properties, such as sintered density [30], atomization energies [31], band gaps [32,33], and phase transitions [34,35]. ...
... Recently, machine learning algorithms and statistical inference [90] have often been used in combination. They are mainly used to predict various properties of material systems effectively and accurately [91]. In the future, they may build material knowledge bases through machine learning and statistical inference [92]. In addition, machine learning methods can be combined with various intelligent algorithms, such as genetic algorithms (GA) [77], which are mainly used to optimize the parameters of machine learning models. Tzuc et al. [93] developed a genetic programming model for optimizing zeolite materials adsorption. Furthermore, GA can explore large and complex search spaces very efficiently [94]. When the amount of data is limited, data mining [95] methods can be used to determine the significant correlations between the ab initio energies of different structures in different materials [2,21,29]. Recently, the method of material data mining has developed rapidly [13] and has produced an open-source toolkit for materials data mining called Matminer [96]. Lu et al. [97] have used data mining to design layered double hydroxides with the required specific surface area. ...
1
2017
... Recently, material discovery and design using machine learning have received increasing attention and have been greatly improved from the standpoints of time efficiency and prediction accuracy [20,21]. Voyles [22] used machine learning algorithms to improve the quality of material microscope data and extract material information from enhanced data. Raccuglia et al. [23] used the support vector machine (SVM) approach to predict the feasibility of untested responses to data collected in failed experiments. Wicker et al. [24] used the SVM algorithm with an RBF kernel to predict the crystallinity of molecular materials. Rupp [25] found that combining quantum mechanics (QM) with machine learning could be expected to improve the accuracy of QM. Stanev et al. [26] proposed that a machine learning model could simulate the critical temperature of superconductors. A neural network can represent high-dimensional potential energy surfaces when the atomistic materials are simulated, and machine-learned interpolation of atomic potential energy surfaces can automatically construct high-precision atomic interaction potentials [[27], [28], [29]]. The predictive power of machine learning algorithms was also reflected in many aspects of material properties, such as sintered density [30], atomization energies [31], band gaps [32,33], and phase transitions [34,35]. ...
2
2016
... Recently, material discovery and design using machine learning have received increasing attention and have been greatly improved from the standpoints of time efficiency and prediction accuracy [20,21]. Voyles [22] used machine learning algorithms to improve the quality of material microscope data and extract material information from enhanced data. Raccuglia et al. [23] used the support vector machine (SVM) approach to predict the feasibility of untested responses to data collected in failed experiments. Wicker et al. [24] used the SVM algorithm with an RBF kernel to predict the crystallinity of molecular materials. Rupp [25] found that combining quantum mechanics (QM) with machine learning could be expected to improve the accuracy of QM. Stanev et al. [26] proposed that a machine learning model could simulate the critical temperature of superconductors. A neural network can represent high-dimensional potential energy surfaces when the atomistic materials are simulated, and machine-learned interpolation of atomic potential energy surfaces can automatically construct high-precision atomic interaction potentials [[27], [28], [29]]. The predictive power of machine learning algorithms was also reflected in many aspects of material properties, such as sintered density [30], atomization energies [31], band gaps [32,33], and phase transitions [34,35]. ...
... Data can come from calculations [41], literature data [42], hypotheses, experiments, and even from failed experiments [23]. Besides, new data can also generate by interpolation of existing data [43]. The amount of material science data is not particularly large, but it is challenging to explore the diversity of data types and the complexity of objects [44]. ...
1
2015
... Recently, material discovery and design using machine learning have received increasing attention and have been greatly improved from the standpoints of time efficiency and prediction accuracy [20,21]. Voyles [22] used machine learning algorithms to improve the quality of material microscope data and extract material information from enhanced data. Raccuglia et al. [23] used the support vector machine (SVM) approach to predict the feasibility of untested responses to data collected in failed experiments. Wicker et al. [24] used the SVM algorithm with an RBF kernel to predict the crystallinity of molecular materials. Rupp [25] found that combining quantum mechanics (QM) with machine learning could be expected to improve the accuracy of QM. Stanev et al. [26] proposed that a machine learning model could simulate the critical temperature of superconductors. A neural network can represent high-dimensional potential energy surfaces when the atomistic materials are simulated, and machine-learned interpolation of atomic potential energy surfaces can automatically construct high-precision atomic interaction potentials [[27], [28], [29]]. The predictive power of machine learning algorithms was also reflected in many aspects of material properties, such as sintered density [30], atomization energies [31], band gaps [32,33], and phase transitions [34,35]. ...
2
2015
... Recently, material discovery and design using machine learning have received increasing attention and have been greatly improved from the standpoints of time efficiency and prediction accuracy [20,21]. Voyles [22] used machine learning algorithms to improve the quality of material microscope data and extract material information from enhanced data. Raccuglia et al. [23] used the support vector machine (SVM) approach to predict the feasibility of untested responses to data collected in failed experiments. Wicker et al. [24] used the SVM algorithm with an RBF kernel to predict the crystallinity of molecular materials. Rupp [25] found that combining quantum mechanics (QM) with machine learning could be expected to improve the accuracy of QM. Stanev et al. [26] proposed that a machine learning model could simulate the critical temperature of superconductors. A neural network can represent high-dimensional potential energy surfaces when the atomistic materials are simulated, and machine-learned interpolation of atomic potential energy surfaces can automatically construct high-precision atomic interaction potentials [[27], [28], [29]]. The predictive power of machine learning algorithms was also reflected in many aspects of material properties, such as sintered density [30], atomization energies [31], band gaps [32,33], and phase transitions [34,35]. ...
... Lilienfeld et al. [131] introduced a machine learning model based on nuclear charges and atomic positions to quickly and accurately predict the atomization energy of various organic molecules and proved its applicability for prediction of molecular atomization potential energy curves. Hansen et al. [132] used machine learning to predict molecular atomization energies, which can significantly accelerate the calculation of quantum chemical properties while maintaining high prediction accuracy. Rupp [25] used kernel ridge regression to predict the atomization energies of small organic molecules. Hansen et al. [133] used machine learning (the so-called Bag of Bonds model; BoB) to estimate molecular atomization energies and predict the exact electronic properties of molecules, Fig. 5 shows that BoB is a stronger machine learning model with proper regularization and improved accuracy by simply expanding the molecular database. Moreover, Pilania et al. [33] and Isayev et al. [134] predicted the bandgap energy. ...
1
2018
... Recently, material discovery and design using machine learning have received increasing attention and have been greatly improved from the standpoints of time efficiency and prediction accuracy [20,21]. Voyles [22] used machine learning algorithms to improve the quality of material microscope data and extract material information from enhanced data. Raccuglia et al. [23] used the support vector machine (SVM) approach to predict the feasibility of untested responses to data collected in failed experiments. Wicker et al. [24] used the SVM algorithm with an RBF kernel to predict the crystallinity of molecular materials. Rupp [25] found that combining quantum mechanics (QM) with machine learning could be expected to improve the accuracy of QM. Stanev et al. [26] proposed that a machine learning model could simulate the critical temperature of superconductors. A neural network can represent high-dimensional potential energy surfaces when the atomistic materials are simulated, and machine-learned interpolation of atomic potential energy surfaces can automatically construct high-precision atomic interaction potentials [[27], [28], [29]]. The predictive power of machine learning algorithms was also reflected in many aspects of material properties, such as sintered density [30], atomization energies [31], band gaps [32,33], and phase transitions [34,35]. ...
1
2007
... Recently, material discovery and design using machine learning have received increasing attention and have been greatly improved from the standpoints of time efficiency and prediction accuracy [20,21]. Voyles [22] used machine learning algorithms to improve the quality of material microscope data and extract material information from enhanced data. Raccuglia et al. [23] used the support vector machine (SVM) approach to predict the feasibility of untested responses to data collected in failed experiments. Wicker et al. [24] used the SVM algorithm with an RBF kernel to predict the crystallinity of molecular materials. Rupp [25] found that combining quantum mechanics (QM) with machine learning could be expected to improve the accuracy of QM. Stanev et al. [26] proposed that a machine learning model could simulate the critical temperature of superconductors. A neural network can represent high-dimensional potential energy surfaces when the atomistic materials are simulated, and machine-learned interpolation of atomic potential energy surfaces can automatically construct high-precision atomic interaction potentials [[27], [28], [29]]. The predictive power of machine learning algorithms was also reflected in many aspects of material properties, such as sintered density [30], atomization energies [31], band gaps [32,33], and phase transitions [34,35]. ...
1
2016
... Recently, material discovery and design using machine learning have received increasing attention and have been greatly improved from the standpoints of time efficiency and prediction accuracy [20,21]. Voyles [22] used machine learning algorithms to improve the quality of material microscope data and extract material information from enhanced data. Raccuglia et al. [23] used the support vector machine (SVM) approach to predict the feasibility of untested responses to data collected in failed experiments. Wicker et al. [24] used the SVM algorithm with an RBF kernel to predict the crystallinity of molecular materials. Rupp [25] found that combining quantum mechanics (QM) with machine learning could be expected to improve the accuracy of QM. Stanev et al. [26] proposed that a machine learning model could simulate the critical temperature of superconductors. A neural network can represent high-dimensional potential energy surfaces when the atomistic materials are simulated, and machine-learned interpolation of atomic potential energy surfaces can automatically construct high-precision atomic interaction potentials [[27], [28], [29]]. The predictive power of machine learning algorithms was also reflected in many aspects of material properties, such as sintered density [30], atomization energies [31], band gaps [32,33], and phase transitions [34,35]. ...
2
2004
... Recently, material discovery and design using machine learning have received increasing attention and have been greatly improved from the standpoints of time efficiency and prediction accuracy [20,21]. Voyles [22] used machine learning algorithms to improve the quality of material microscope data and extract material information from enhanced data. Raccuglia et al. [23] used the support vector machine (SVM) approach to predict the feasibility of untested responses to data collected in failed experiments. Wicker et al. [24] used the SVM algorithm with an RBF kernel to predict the crystallinity of molecular materials. Rupp [25] found that combining quantum mechanics (QM) with machine learning could be expected to improve the accuracy of QM. Stanev et al. [26] proposed that a machine learning model could simulate the critical temperature of superconductors. A neural network can represent high-dimensional potential energy surfaces when the atomistic materials are simulated, and machine-learned interpolation of atomic potential energy surfaces can automatically construct high-precision atomic interaction potentials [[27], [28], [29]]. The predictive power of machine learning algorithms was also reflected in many aspects of material properties, such as sintered density [30], atomization energies [31], band gaps [32,33], and phase transitions [34,35]. ...
... Recently, machine learning algorithms and statistical inference [90] have often been used in combination. They are mainly used to predict various properties of material systems effectively and accurately [91]. In the future, they may build material knowledge bases through machine learning and statistical inference [92]. In addition, machine learning methods can be combined with various intelligent algorithms, such as genetic algorithms (GA) [77], which are mainly used to optimize the parameters of machine learning models. Tzuc et al. [93] developed a genetic programming model for optimizing zeolite materials adsorption. Furthermore, GA can explore large and complex search spaces very efficiently [94]. When the amount of data is limited, data mining [95] methods can be used to determine the significant correlations between the ab initio energies of different structures in different materials [2,21,29]. Recently, the method of material data mining has developed rapidly [13] and has produced an open-source toolkit for materials data mining called Matminer [96]. Lu et al. [97] have used data mining to design layered double hydroxides with the required specific surface area. ...
1
2018
... Recently, material discovery and design using machine learning have received increasing attention and have been greatly improved from the standpoints of time efficiency and prediction accuracy [20,21]. Voyles [22] used machine learning algorithms to improve the quality of material microscope data and extract material information from enhanced data. Raccuglia et al. [23] used the support vector machine (SVM) approach to predict the feasibility of untested responses to data collected in failed experiments. Wicker et al. [24] used the SVM algorithm with an RBF kernel to predict the crystallinity of molecular materials. Rupp [25] found that combining quantum mechanics (QM) with machine learning could be expected to improve the accuracy of QM. Stanev et al. [26] proposed that a machine learning model could simulate the critical temperature of superconductors. A neural network can represent high-dimensional potential energy surfaces when the atomistic materials are simulated, and machine-learned interpolation of atomic potential energy surfaces can automatically construct high-precision atomic interaction potentials [[27], [28], [29]]. The predictive power of machine learning algorithms was also reflected in many aspects of material properties, such as sintered density [30], atomization energies [31], band gaps [32,33], and phase transitions [34,35]. ...
2
2015
... Recently, material discovery and design using machine learning have received increasing attention and have been greatly improved from the standpoints of time efficiency and prediction accuracy [20,21]. Voyles [22] used machine learning algorithms to improve the quality of material microscope data and extract material information from enhanced data. Raccuglia et al. [23] used the support vector machine (SVM) approach to predict the feasibility of untested responses to data collected in failed experiments. Wicker et al. [24] used the SVM algorithm with an RBF kernel to predict the crystallinity of molecular materials. Rupp [25] found that combining quantum mechanics (QM) with machine learning could be expected to improve the accuracy of QM. Stanev et al. [26] proposed that a machine learning model could simulate the critical temperature of superconductors. A neural network can represent high-dimensional potential energy surfaces when the atomistic materials are simulated, and machine-learned interpolation of atomic potential energy surfaces can automatically construct high-precision atomic interaction potentials [[27], [28], [29]]. The predictive power of machine learning algorithms was also reflected in many aspects of material properties, such as sintered density [30], atomization energies [31], band gaps [32,33], and phase transitions [34,35]. ...
... Machine learning should be viewed as the sum of the organization of the initial datasets, the descriptor creation and algorithm learning steps, and the necessary subsequent steps for targeted new data entry. Ultimately, the expert recommendation system can be improved continuously and adaptively [43]. Material data are made up of datasets, and dataset uncertainties also have a positive impact on machine learning model predictions [75]. Therefore, Bhat et al. [76] established a set of general rules for describing material data that will aid machine learning and decision-making. Material performance can be predicted through machine learning. In other words, the material is first “fingerprinted” (also called generating a descriptor or a feature vector), followed by a mapping between the descriptor and the property of interest, and finally performance prediction [31,43]. No matter what kind of material is studied, the condition of machine learning is the existence of past data. In machine learning algorithms, the input is generally a component or process parameter of the material, and the output is generally the property or properties of interest [77]. Fig. 1 shows the specific process of machine learning [43]. In Fig. 1, N and M are respectively the number of training examples and the number of fingerprint components. ...
1
2016
... Recently, material discovery and design using machine learning have received increasing attention and have been greatly improved from the standpoints of time efficiency and prediction accuracy [20,21]. Voyles [22] used machine learning algorithms to improve the quality of material microscope data and extract material information from enhanced data. Raccuglia et al. [23] used the support vector machine (SVM) approach to predict the feasibility of untested responses to data collected in failed experiments. Wicker et al. [24] used the SVM algorithm with an RBF kernel to predict the crystallinity of molecular materials. Rupp [25] found that combining quantum mechanics (QM) with machine learning could be expected to improve the accuracy of QM. Stanev et al. [26] proposed that a machine learning model could simulate the critical temperature of superconductors. A neural network can represent high-dimensional potential energy surfaces when the atomistic materials are simulated, and machine-learned interpolation of atomic potential energy surfaces can automatically construct high-precision atomic interaction potentials [[27], [28], [29]]. The predictive power of machine learning algorithms was also reflected in many aspects of material properties, such as sintered density [30], atomization energies [31], band gaps [32,33], and phase transitions [34,35]. ...
2
2016
... Recently, material discovery and design using machine learning have received increasing attention and have been greatly improved from the standpoints of time efficiency and prediction accuracy [20,21]. Voyles [22] used machine learning algorithms to improve the quality of material microscope data and extract material information from enhanced data. Raccuglia et al. [23] used the support vector machine (SVM) approach to predict the feasibility of untested responses to data collected in failed experiments. Wicker et al. [24] used the SVM algorithm with an RBF kernel to predict the crystallinity of molecular materials. Rupp [25] found that combining quantum mechanics (QM) with machine learning could be expected to improve the accuracy of QM. Stanev et al. [26] proposed that a machine learning model could simulate the critical temperature of superconductors. A neural network can represent high-dimensional potential energy surfaces when the atomistic materials are simulated, and machine-learned interpolation of atomic potential energy surfaces can automatically construct high-precision atomic interaction potentials [[27], [28], [29]]. The predictive power of machine learning algorithms was also reflected in many aspects of material properties, such as sintered density [30], atomization energies [31], band gaps [32,33], and phase transitions [34,35]. ...
... Lilienfeld et al. [131] introduced a machine learning model based on nuclear charges and atomic positions to quickly and accurately predict the atomization energy of various organic molecules and proved its applicability for prediction of molecular atomization potential energy curves. Hansen et al. [132] used machine learning to predict molecular atomization energies, which can significantly accelerate the calculation of quantum chemical properties while maintaining high prediction accuracy. Rupp [25] used kernel ridge regression to predict the atomization energies of small organic molecules. Hansen et al. [133] used machine learning (the so-called Bag of Bonds model; BoB) to estimate molecular atomization energies and predict the exact electronic properties of molecules, Fig. 5 shows that BoB is a stronger machine learning model with proper regularization and improved accuracy by simply expanding the molecular database. Moreover, Pilania et al. [33] and Isayev et al. [134] predicted the bandgap energy. ...
1
2008
... Recently, material discovery and design using machine learning have received increasing attention and have been greatly improved from the standpoints of time efficiency and prediction accuracy [20,21]. Voyles [22] used machine learning algorithms to improve the quality of material microscope data and extract material information from enhanced data. Raccuglia et al. [23] used the support vector machine (SVM) approach to predict the feasibility of untested responses to data collected in failed experiments. Wicker et al. [24] used the SVM algorithm with an RBF kernel to predict the crystallinity of molecular materials. Rupp [25] found that combining quantum mechanics (QM) with machine learning could be expected to improve the accuracy of QM. Stanev et al. [26] proposed that a machine learning model could simulate the critical temperature of superconductors. A neural network can represent high-dimensional potential energy surfaces when the atomistic materials are simulated, and machine-learned interpolation of atomic potential energy surfaces can automatically construct high-precision atomic interaction potentials [[27], [28], [29]]. The predictive power of machine learning algorithms was also reflected in many aspects of material properties, such as sintered density [30], atomization energies [31], band gaps [32,33], and phase transitions [34,35]. ...
1
2008
... Recently, material discovery and design using machine learning have received increasing attention and have been greatly improved from the standpoints of time efficiency and prediction accuracy [20,21]. Voyles [22] used machine learning algorithms to improve the quality of material microscope data and extract material information from enhanced data. Raccuglia et al. [23] used the support vector machine (SVM) approach to predict the feasibility of untested responses to data collected in failed experiments. Wicker et al. [24] used the SVM algorithm with an RBF kernel to predict the crystallinity of molecular materials. Rupp [25] found that combining quantum mechanics (QM) with machine learning could be expected to improve the accuracy of QM. Stanev et al. [26] proposed that a machine learning model could simulate the critical temperature of superconductors. A neural network can represent high-dimensional potential energy surfaces when the atomistic materials are simulated, and machine-learned interpolation of atomic potential energy surfaces can automatically construct high-precision atomic interaction potentials [[27], [28], [29]]. The predictive power of machine learning algorithms was also reflected in many aspects of material properties, such as sintered density [30], atomization energies [31], band gaps [32,33], and phase transitions [34,35]. ...
1
2014
... Continuous advancement in science depends on shared and repeatable data [36]. Large-scale data analysis is the driving force for material progress. Materials science is on the verge of adopting Materials 4.0 /fourth paradigm [37,38] or data-driven [39] discovery on large-scale data [40]. The quality of data analysis and data mining is affected by the data quality of the materials database and directly affects the application of data that are mined. The existing database is far from including all known materials, not to mention all possible materials. Data-driven discovery applies to certain materials, but not to all materials. Even if people pick an interesting material on their computers, it will take years to synthesize it in the laboratory [1]. ...
1
2018
... Continuous advancement in science depends on shared and repeatable data [36]. Large-scale data analysis is the driving force for material progress. Materials science is on the verge of adopting Materials 4.0 /fourth paradigm [37,38] or data-driven [39] discovery on large-scale data [40]. The quality of data analysis and data mining is affected by the data quality of the materials database and directly affects the application of data that are mined. The existing database is far from including all known materials, not to mention all possible materials. Data-driven discovery applies to certain materials, but not to all materials. Even if people pick an interesting material on their computers, it will take years to synthesize it in the laboratory [1]. ...
1
2016
... Continuous advancement in science depends on shared and repeatable data [36]. Large-scale data analysis is the driving force for material progress. Materials science is on the verge of adopting Materials 4.0 /fourth paradigm [37,38] or data-driven [39] discovery on large-scale data [40]. The quality of data analysis and data mining is affected by the data quality of the materials database and directly affects the application of data that are mined. The existing database is far from including all known materials, not to mention all possible materials. Data-driven discovery applies to certain materials, but not to all materials. Even if people pick an interesting material on their computers, it will take years to synthesize it in the laboratory [1]. ...
1
2017
... Continuous advancement in science depends on shared and repeatable data [36]. Large-scale data analysis is the driving force for material progress. Materials science is on the verge of adopting Materials 4.0 /fourth paradigm [37,38] or data-driven [39] discovery on large-scale data [40]. The quality of data analysis and data mining is affected by the data quality of the materials database and directly affects the application of data that are mined. The existing database is far from including all known materials, not to mention all possible materials. Data-driven discovery applies to certain materials, but not to all materials. Even if people pick an interesting material on their computers, it will take years to synthesize it in the laboratory [1]. ...
1
2017
... Continuous advancement in science depends on shared and repeatable data [36]. Large-scale data analysis is the driving force for material progress. Materials science is on the verge of adopting Materials 4.0 /fourth paradigm [37,38] or data-driven [39] discovery on large-scale data [40]. The quality of data analysis and data mining is affected by the data quality of the materials database and directly affects the application of data that are mined. The existing database is far from including all known materials, not to mention all possible materials. Data-driven discovery applies to certain materials, but not to all materials. Even if people pick an interesting material on their computers, it will take years to synthesize it in the laboratory [1]. ...
1
2017
... Data can come from calculations [41], literature data [42], hypotheses, experiments, and even from failed experiments [23]. Besides, new data can also generate by interpolation of existing data [43]. The amount of material science data is not particularly large, but it is challenging to explore the diversity of data types and the complexity of objects [44]. ...
3
2017
... Data can come from calculations [41], literature data [42], hypotheses, experiments, and even from failed experiments [23]. Besides, new data can also generate by interpolation of existing data [43]. The amount of material science data is not particularly large, but it is challenging to explore the diversity of data types and the complexity of objects [44]. ...
... In addition, there are the computational materials repository [61], computational materials property databases (www.synthesisproject.org) [42], MatNavi materials databases [62], the materials data facility [63], interatomic potential (force field) databases [64], the Superconducting Material Database (SuperCon) [65], the structural materials database (The Materials Commons) [66], and many others. ...
... At present, the scale of data that can be applied to machine learning in the field of materials is still very small, making it impossible to generate rigorous predictions of material properties and new material discoveries through machine learning. The predicted results are often only close to the probability values of real data and cannot be truly used for experimental guidance. Even if some experts propose to improve machine learning application methods for small-scale datasets, for example by adding a rough attribute estimation to the feature space to build a machine learning model using a small-scale material dataset, the accuracy of the prediction can be improved [153]. However, even with the gradual development of machine learning and deep learning algorithms, from the application experience of each algorithm model, the scale of the dataset is still the main factor in model prediction accuracy. This problem is reflected not only in the machine learning algorithms, but also in deep learning, which has attracted much attention in recent years. In the last few decades, it has been found that material data are presented relatively intact in a large number of scientific papers, and literature data extraction has made some progress in many specific fields, such as chemistry [154] and biomedicine [[155], [156], [157]]. Some studies using these material data have already been carried out. For example, a paper information-assisted extraction software package has been developed and is available online (https://www.mgedata.cn/) [158]. Xie et al. [159] designed the composition of high-performance copper alloys using machine learning based on data from the literature. Edward et al. [42] obtained material synthesis information from academic publications. Kononova et al. [160] obtained inorganic material synthesis recipes by using text mining of scientific publications. However, data in the literature are rarely used by material scientists, and even if the data are used, the data scale is small because there is no mature method to extract data from the material literature. So far, no relatively complete corpus has been developed in the materials field to support the application of material data. Therefore, in the materials genome initiative, material prediction through machine learning should not only focus on research into the machine learning algorithm itself, but should also extract valuable material data from the materials science literature. This would involve first classifying and pre-processing these literature data, then using machine learning algorithms to predict the performance data of interest, and finally finding the correlations among material composition, process, structure, and performance. These steps will play a key role in discovering new properties of materials. ...
5
2017
... Data can come from calculations [41], literature data [42], hypotheses, experiments, and even from failed experiments [23]. Besides, new data can also generate by interpolation of existing data [43]. The amount of material science data is not particularly large, but it is challenging to explore the diversity of data types and the complexity of objects [44]. ...
... Machine learning should be viewed as the sum of the organization of the initial datasets, the descriptor creation and algorithm learning steps, and the necessary subsequent steps for targeted new data entry. Ultimately, the expert recommendation system can be improved continuously and adaptively [43]. Material data are made up of datasets, and dataset uncertainties also have a positive impact on machine learning model predictions [75]. Therefore, Bhat et al. [76] established a set of general rules for describing material data that will aid machine learning and decision-making. Material performance can be predicted through machine learning. In other words, the material is first “fingerprinted” (also called generating a descriptor or a feature vector), followed by a mapping between the descriptor and the property of interest, and finally performance prediction [31,43]. No matter what kind of material is studied, the condition of machine learning is the existence of past data. In machine learning algorithms, the input is generally a component or process parameter of the material, and the output is generally the property or properties of interest [77]. Fig. 1 shows the specific process of machine learning [43]. In Fig. 1, N and M are respectively the number of training examples and the number of fingerprint components. ...
... ,43]. No matter what kind of material is studied, the condition of machine learning is the existence of past data. In machine learning algorithms, the input is generally a component or process parameter of the material, and the output is generally the property or properties of interest [77]. Fig. 1 shows the specific process of machine learning [43]. In Fig. 1, N and M are respectively the number of training examples and the number of fingerprint components. ...
... shows the specific process of machine learning [43]. In Fig. 1, N and M are respectively the number of training examples and the number of fingerprint components. ...
... The process of specific machine learning in materials science. (a) Schematic view of an example data set; (b) statement of the learning problem; (c) creation of a surrogate prediction model via the fingerprinting and learning steps [43].
As shown in Fig. 2, machine learning algorithms commonly used in materials science can be divided into four categories: probability estimation [20], regression, clustering, and classification [78]. The main machine learning methods for material performance prediction are regression, clustering, and classification algorithms [20]. Examples include kernel ridge regression [79], artificial neural networks (ANN) [[80], [81], [82]], SVM [83], Gaussian process regression [84], feature learning, clustering, matrix factorization, and constraint reasoning [85], Adaboost [86], and decision trees [87]. Machine learning can also be used to discover new materials, for example, using probability estimation algorithms [88] or principal component analysis [2,89]. ...
1
2016
... Data can come from calculations [41], literature data [42], hypotheses, experiments, and even from failed experiments [23]. Besides, new data can also generate by interpolation of existing data [43]. The amount of material science data is not particularly large, but it is challenging to explore the diversity of data types and the complexity of objects [44]. ...
1
1983
... An early database, The Inorganic Crystal Structure Database [45], created in 2002, which covers the literature from 1915 to 2002, is a comprehensive collection of crystal structure information for non-organic compounds, including inorganics, minerals, ceramics, and metals [46]. In 2015, Wolverton [47] launched the Open Quantum Materials Database (OQMD), which is also an inorganic crystal structure database with approximately 50,000 pieces of data from a widely used experimental library [1]. Cambridge Structural Databases (CSD) [48] is a worldwide repository for small-molecule organic and metal-organic crystal structures hosted by Cambridge Crystallographic Data Center (CCDC) (https://www.lib.ncsu.edu/databases/cambridge-structural.html [49]). The Materials Projec t [50,51] is the core program of the Materials Genome Initiative, which uses high-throughput calculations to reveal the properties of all known inorganic materials. These open datasets can be accessed through multiple channels for interactive exploration and data mining. The Materials Project URL is https://www.materialsproject.org [52]. By September 2019, the database contained 120,612 inorganic compounds, 52,366 band structures, 35,336 molecules, 530,243 nanoporous materials, 13,621 elastic tensors, 3003 piezoelectric tensors, 4401 intercalation electrodes, and 16,128 conversion electrodes. ...
1
2002
... An early database, The Inorganic Crystal Structure Database [45], created in 2002, which covers the literature from 1915 to 2002, is a comprehensive collection of crystal structure information for non-organic compounds, including inorganics, minerals, ceramics, and metals [46]. In 2015, Wolverton [47] launched the Open Quantum Materials Database (OQMD), which is also an inorganic crystal structure database with approximately 50,000 pieces of data from a widely used experimental library [1]. Cambridge Structural Databases (CSD) [48] is a worldwide repository for small-molecule organic and metal-organic crystal structures hosted by Cambridge Crystallographic Data Center (CCDC) (https://www.lib.ncsu.edu/databases/cambridge-structural.html [49]). The Materials Projec t [50,51] is the core program of the Materials Genome Initiative, which uses high-throughput calculations to reveal the properties of all known inorganic materials. These open datasets can be accessed through multiple channels for interactive exploration and data mining. The Materials Project URL is https://www.materialsproject.org [52]. By September 2019, the database contained 120,612 inorganic compounds, 52,366 band structures, 35,336 molecules, 530,243 nanoporous materials, 13,621 elastic tensors, 3003 piezoelectric tensors, 4401 intercalation electrodes, and 16,128 conversion electrodes. ...
2
2015
... An early database, The Inorganic Crystal Structure Database [45], created in 2002, which covers the literature from 1915 to 2002, is a comprehensive collection of crystal structure information for non-organic compounds, including inorganics, minerals, ceramics, and metals [46]. In 2015, Wolverton [47] launched the Open Quantum Materials Database (OQMD), which is also an inorganic crystal structure database with approximately 50,000 pieces of data from a widely used experimental library [1]. Cambridge Structural Databases (CSD) [48] is a worldwide repository for small-molecule organic and metal-organic crystal structures hosted by Cambridge Crystallographic Data Center (CCDC) (https://www.lib.ncsu.edu/databases/cambridge-structural.html [49]). The Materials Projec t [50,51] is the core program of the Materials Genome Initiative, which uses high-throughput calculations to reveal the properties of all known inorganic materials. These open datasets can be accessed through multiple channels for interactive exploration and data mining. The Materials Project URL is https://www.materialsproject.org [52]. By September 2019, the database contained 120,612 inorganic compounds, 52,366 band structures, 35,336 molecules, 530,243 nanoporous materials, 13,621 elastic tensors, 3003 piezoelectric tensors, 4401 intercalation electrodes, and 16,128 conversion electrodes. ...
... In 2003, Ceder et al. [1] first demonstrated how quantum mechanical computing databases could help predict the most likely crystal structure of metal alloys—a crucial step in the invention of new materials. Ceder introduced the concept of public libraries of material properties and used data mining to fill in missing data. In the second year after the genome project was launched, Curtarolo [53] posted a database based on his software, called AFLOWlib, which is a database with more than one million materials and approximately 100 million calculated properties. AFLOWlib contains such a large amount of data because it also includes thousands of hypothetical materials, many of which exist for only one second in the real world, but Curtarolo used these data to study why some alloys can form metallic glass [54]. OQMD is also a high-throughput database that includes a large number of hypothetical materials, and the entire database can be obtained for free at http://www.oqmd.org/download [47]. ...
1
2002
... An early database, The Inorganic Crystal Structure Database [45], created in 2002, which covers the literature from 1915 to 2002, is a comprehensive collection of crystal structure information for non-organic compounds, including inorganics, minerals, ceramics, and metals [46]. In 2015, Wolverton [47] launched the Open Quantum Materials Database (OQMD), which is also an inorganic crystal structure database with approximately 50,000 pieces of data from a widely used experimental library [1]. Cambridge Structural Databases (CSD) [48] is a worldwide repository for small-molecule organic and metal-organic crystal structures hosted by Cambridge Crystallographic Data Center (CCDC) (https://www.lib.ncsu.edu/databases/cambridge-structural.html [49]). The Materials Projec t [50,51] is the core program of the Materials Genome Initiative, which uses high-throughput calculations to reveal the properties of all known inorganic materials. These open datasets can be accessed through multiple channels for interactive exploration and data mining. The Materials Project URL is https://www.materialsproject.org [52]. By September 2019, the database contained 120,612 inorganic compounds, 52,366 band structures, 35,336 molecules, 530,243 nanoporous materials, 13,621 elastic tensors, 3003 piezoelectric tensors, 4401 intercalation electrodes, and 16,128 conversion electrodes. ...
1
2019
... An early database, The Inorganic Crystal Structure Database [45], created in 2002, which covers the literature from 1915 to 2002, is a comprehensive collection of crystal structure information for non-organic compounds, including inorganics, minerals, ceramics, and metals [46]. In 2015, Wolverton [47] launched the Open Quantum Materials Database (OQMD), which is also an inorganic crystal structure database with approximately 50,000 pieces of data from a widely used experimental library [1]. Cambridge Structural Databases (CSD) [48] is a worldwide repository for small-molecule organic and metal-organic crystal structures hosted by Cambridge Crystallographic Data Center (CCDC) (https://www.lib.ncsu.edu/databases/cambridge-structural.html [49]). The Materials Projec t [50,51] is the core program of the Materials Genome Initiative, which uses high-throughput calculations to reveal the properties of all known inorganic materials. These open datasets can be accessed through multiple channels for interactive exploration and data mining. The Materials Project URL is https://www.materialsproject.org [52]. By September 2019, the database contained 120,612 inorganic compounds, 52,366 band structures, 35,336 molecules, 530,243 nanoporous materials, 13,621 elastic tensors, 3003 piezoelectric tensors, 4401 intercalation electrodes, and 16,128 conversion electrodes. ...
1
2013
... An early database, The Inorganic Crystal Structure Database [45], created in 2002, which covers the literature from 1915 to 2002, is a comprehensive collection of crystal structure information for non-organic compounds, including inorganics, minerals, ceramics, and metals [46]. In 2015, Wolverton [47] launched the Open Quantum Materials Database (OQMD), which is also an inorganic crystal structure database with approximately 50,000 pieces of data from a widely used experimental library [1]. Cambridge Structural Databases (CSD) [48] is a worldwide repository for small-molecule organic and metal-organic crystal structures hosted by Cambridge Crystallographic Data Center (CCDC) (https://www.lib.ncsu.edu/databases/cambridge-structural.html [49]). The Materials Projec t [50,51] is the core program of the Materials Genome Initiative, which uses high-throughput calculations to reveal the properties of all known inorganic materials. These open datasets can be accessed through multiple channels for interactive exploration and data mining. The Materials Project URL is https://www.materialsproject.org [52]. By September 2019, the database contained 120,612 inorganic compounds, 52,366 band structures, 35,336 molecules, 530,243 nanoporous materials, 13,621 elastic tensors, 3003 piezoelectric tensors, 4401 intercalation electrodes, and 16,128 conversion electrodes. ...
2
2017
... An early database, The Inorganic Crystal Structure Database [45], created in 2002, which covers the literature from 1915 to 2002, is a comprehensive collection of crystal structure information for non-organic compounds, including inorganics, minerals, ceramics, and metals [46]. In 2015, Wolverton [47] launched the Open Quantum Materials Database (OQMD), which is also an inorganic crystal structure database with approximately 50,000 pieces of data from a widely used experimental library [1]. Cambridge Structural Databases (CSD) [48] is a worldwide repository for small-molecule organic and metal-organic crystal structures hosted by Cambridge Crystallographic Data Center (CCDC) (https://www.lib.ncsu.edu/databases/cambridge-structural.html [49]). The Materials Projec t [50,51] is the core program of the Materials Genome Initiative, which uses high-throughput calculations to reveal the properties of all known inorganic materials. These open datasets can be accessed through multiple channels for interactive exploration and data mining. The Materials Project URL is https://www.materialsproject.org [52]. By September 2019, the database contained 120,612 inorganic compounds, 52,366 band structures, 35,336 molecules, 530,243 nanoporous materials, 13,621 elastic tensors, 3003 piezoelectric tensors, 4401 intercalation electrodes, and 16,128 conversion electrodes. ...
... To accelerate the design of new materials, recent studies have screened out many types of promising candidate materials [51], including perovskite compounds [83,142], metal-organic frameworks [143,144], catalysts [145,146], light-emitting molecules [147], and thermoelectric materials [[148], [149], [150], [151]]. A large-scale database based on quantum mechanical calculation of known materials, using machine learning to calculate material screening, is a combination screening process for new materials in unconstrained component space, which can be used for an extensive range of critical material discoveries [87]. In addition, interface structure and energy can be also predicted through virtual screening [152]. ...
1
2019
... An early database, The Inorganic Crystal Structure Database [45], created in 2002, which covers the literature from 1915 to 2002, is a comprehensive collection of crystal structure information for non-organic compounds, including inorganics, minerals, ceramics, and metals [46]. In 2015, Wolverton [47] launched the Open Quantum Materials Database (OQMD), which is also an inorganic crystal structure database with approximately 50,000 pieces of data from a widely used experimental library [1]. Cambridge Structural Databases (CSD) [48] is a worldwide repository for small-molecule organic and metal-organic crystal structures hosted by Cambridge Crystallographic Data Center (CCDC) (https://www.lib.ncsu.edu/databases/cambridge-structural.html [49]). The Materials Projec t [50,51] is the core program of the Materials Genome Initiative, which uses high-throughput calculations to reveal the properties of all known inorganic materials. These open datasets can be accessed through multiple channels for interactive exploration and data mining. The Materials Project URL is https://www.materialsproject.org [52]. By September 2019, the database contained 120,612 inorganic compounds, 52,366 band structures, 35,336 molecules, 530,243 nanoporous materials, 13,621 elastic tensors, 3003 piezoelectric tensors, 4401 intercalation electrodes, and 16,128 conversion electrodes. ...
1
2012
... In 2003, Ceder et al. [1] first demonstrated how quantum mechanical computing databases could help predict the most likely crystal structure of metal alloys—a crucial step in the invention of new materials. Ceder introduced the concept of public libraries of material properties and used data mining to fill in missing data. In the second year after the genome project was launched, Curtarolo [53] posted a database based on his software, called AFLOWlib, which is a database with more than one million materials and approximately 100 million calculated properties. AFLOWlib contains such a large amount of data because it also includes thousands of hypothetical materials, many of which exist for only one second in the real world, but Curtarolo used these data to study why some alloys can form metallic glass [54]. OQMD is also a high-throughput database that includes a large number of hypothetical materials, and the entire database can be obtained for free at http://www.oqmd.org/download [47]. ...
1
2018
... In 2003, Ceder et al. [1] first demonstrated how quantum mechanical computing databases could help predict the most likely crystal structure of metal alloys—a crucial step in the invention of new materials. Ceder introduced the concept of public libraries of material properties and used data mining to fill in missing data. In the second year after the genome project was launched, Curtarolo [53] posted a database based on his software, called AFLOWlib, which is a database with more than one million materials and approximately 100 million calculated properties. AFLOWlib contains such a large amount of data because it also includes thousands of hypothetical materials, many of which exist for only one second in the real world, but Curtarolo used these data to study why some alloys can form metallic glass [54]. OQMD is also a high-throughput database that includes a large number of hypothetical materials, and the entire database can be obtained for free at http://www.oqmd.org/download [47]. ...
1
2016
... The maturity of high-throughput first-principles calculations, along with the rise of virtual material screening, has created many accessible databases for the materials science community, including Citrination Platform [55], large molecular libraries [56], and the Harvard Clean Energy Project Database (CEPDB) [57]. ...
1
2015
... The maturity of high-throughput first-principles calculations, along with the rise of virtual material screening, has created many accessible databases for the materials science community, including Citrination Platform [55], large molecular libraries [56], and the Harvard Clean Energy Project Database (CEPDB) [57]. ...
1
2014
... The maturity of high-throughput first-principles calculations, along with the rise of virtual material screening, has created many accessible databases for the materials science community, including Citrination Platform [55], large molecular libraries [56], and the Harvard Clean Energy Project Database (CEPDB) [57]. ...
1
2019
... MatWeb is the world's largest database of material properties, with over 130,000 metals, plastics, ceramics, and composites [58]. Total Materia is the world’s most comprehensive materials database, which is based on 74 international standards and offers more than 15,000,000 materials [59]. ASM International is the world’s largest and most established materials information society, which can connect anyone to a global network of peers and provides access to trusted materials information [60]. ...
1
2019
... MatWeb is the world's largest database of material properties, with over 130,000 metals, plastics, ceramics, and composites [58]. Total Materia is the world’s most comprehensive materials database, which is based on 74 international standards and offers more than 15,000,000 materials [59]. ASM International is the world’s largest and most established materials information society, which can connect anyone to a global network of peers and provides access to trusted materials information [60]. ...
1
2019
... MatWeb is the world's largest database of material properties, with over 130,000 metals, plastics, ceramics, and composites [58]. Total Materia is the world’s most comprehensive materials database, which is based on 74 international standards and offers more than 15,000,000 materials [59]. ASM International is the world’s largest and most established materials information society, which can connect anyone to a global network of peers and provides access to trusted materials information [60]. ...
1
2012
... In addition, there are the computational materials repository [61], computational materials property databases (www.synthesisproject.org) [42], MatNavi materials databases [62], the materials data facility [63], interatomic potential (force field) databases [64], the Superconducting Material Database (SuperCon) [65], the structural materials database (The Materials Commons) [66], and many others. ...
1
2019
... In addition, there are the computational materials repository [61], computational materials property databases (www.synthesisproject.org) [42], MatNavi materials databases [62], the materials data facility [63], interatomic potential (force field) databases [64], the Superconducting Material Database (SuperCon) [65], the structural materials database (The Materials Commons) [66], and many others. ...
1
2016
... In addition, there are the computational materials repository [61], computational materials property databases (www.synthesisproject.org) [42], MatNavi materials databases [62], the materials data facility [63], interatomic potential (force field) databases [64], the Superconducting Material Database (SuperCon) [65], the structural materials database (The Materials Commons) [66], and many others. ...
1
2019
... In addition, there are the computational materials repository [61], computational materials property databases (www.synthesisproject.org) [42], MatNavi materials databases [62], the materials data facility [63], interatomic potential (force field) databases [64], the Superconducting Material Database (SuperCon) [65], the structural materials database (The Materials Commons) [66], and many others. ...
1
2019
... In addition, there are the computational materials repository [61], computational materials property databases (www.synthesisproject.org) [42], MatNavi materials databases [62], the materials data facility [63], interatomic potential (force field) databases [64], the Superconducting Material Database (SuperCon) [65], the structural materials database (The Materials Commons) [66], and many others. ...
1
2016
... In addition, there are the computational materials repository [61], computational materials property databases (www.synthesisproject.org) [42], MatNavi materials databases [62], the materials data facility [63], interatomic potential (force field) databases [64], the Superconducting Material Database (SuperCon) [65], the structural materials database (The Materials Commons) [66], and many others. ...
1
2016
... The Air Force Research Laboratory has developed an integrated collaborative environment that connects laboratory equipment through a laboratory intranet to serve the internal needs of large material laboratories [67]. ...
1
2017
... Microstructure-informed cloud computing provides a practical platform for interoperability of material datasets and data analytics tools [68]. An online computation platform for materials data mining can perform data mining on materials [69]. AiiDA can be used to calculate automatic interactions in materials science [70]. MatCloud is a high-throughput computing infrastructure for the comprehensive management of materials simulation, data, and resources [71]. In addition, the VNL software package provides a graphical environment for first-principles simulations [72]. ...
1
2018
... Microstructure-informed cloud computing provides a practical platform for interoperability of material datasets and data analytics tools [68]. An online computation platform for materials data mining can perform data mining on materials [69]. AiiDA can be used to calculate automatic interactions in materials science [70]. MatCloud is a high-throughput computing infrastructure for the comprehensive management of materials simulation, data, and resources [71]. In addition, the VNL software package provides a graphical environment for first-principles simulations [72]. ...
1
2016
... Microstructure-informed cloud computing provides a practical platform for interoperability of material datasets and data analytics tools [68]. An online computation platform for materials data mining can perform data mining on materials [69]. AiiDA can be used to calculate automatic interactions in materials science [70]. MatCloud is a high-throughput computing infrastructure for the comprehensive management of materials simulation, data, and resources [71]. In addition, the VNL software package provides a graphical environment for first-principles simulations [72]. ...
1
2018
... Microstructure-informed cloud computing provides a practical platform for interoperability of material datasets and data analytics tools [68]. An online computation platform for materials data mining can perform data mining on materials [69]. AiiDA can be used to calculate automatic interactions in materials science [70]. MatCloud is a high-throughput computing infrastructure for the comprehensive management of materials simulation, data, and resources [71]. In addition, the VNL software package provides a graphical environment for first-principles simulations [72]. ...
1
2019
... Microstructure-informed cloud computing provides a practical platform for interoperability of material datasets and data analytics tools [68]. An online computation platform for materials data mining can perform data mining on materials [69]. AiiDA can be used to calculate automatic interactions in materials science [70]. MatCloud is a high-throughput computing infrastructure for the comprehensive management of materials simulation, data, and resources [71]. In addition, the VNL software package provides a graphical environment for first-principles simulations [72]. ...
1
2017
... Big Data in materials science cannot directly see the structure of these data using standard tools. Therefore, it is necessary to discover the underlying laws of data based on new methods [73]. The new era of Big Data science and analytics offers a rather compelling approach: machine learning to help with discovery, design, and optimization of novel materials. The machine learning method has developed rapidly, and its long-term goal is the continuous learning and proficiency of the algorithm. For example, without human knowledge, AlphaGo Zero defeated AlphaGo based on a reinforcement learning algorithm, which proved the importance of the continuous learning and progress of the algorithm [74]. ...
1
2017
... Big Data in materials science cannot directly see the structure of these data using standard tools. Therefore, it is necessary to discover the underlying laws of data based on new methods [73]. The new era of Big Data science and analytics offers a rather compelling approach: machine learning to help with discovery, design, and optimization of novel materials. The machine learning method has developed rapidly, and its long-term goal is the continuous learning and proficiency of the algorithm. For example, without human knowledge, AlphaGo Zero defeated AlphaGo based on a reinforcement learning algorithm, which proved the importance of the continuous learning and progress of the algorithm [74]. ...
2
2019
... Machine learning should be viewed as the sum of the organization of the initial datasets, the descriptor creation and algorithm learning steps, and the necessary subsequent steps for targeted new data entry. Ultimately, the expert recommendation system can be improved continuously and adaptively [43]. Material data are made up of datasets, and dataset uncertainties also have a positive impact on machine learning model predictions [75]. Therefore, Bhat et al. [76] established a set of general rules for describing material data that will aid machine learning and decision-making. Material performance can be predicted through machine learning. In other words, the material is first “fingerprinted” (also called generating a descriptor or a feature vector), followed by a mapping between the descriptor and the property of interest, and finally performance prediction [31,43]. No matter what kind of material is studied, the condition of machine learning is the existence of past data. In machine learning algorithms, the input is generally a component or process parameter of the material, and the output is generally the property or properties of interest [77]. Fig. 1 shows the specific process of machine learning [43]. In Fig. 1, N and M are respectively the number of training examples and the number of fingerprint components. ...
... Other applications exist for macroscopic performance prediction. Seko et al. [124] used four regression algorithms to predict the melting temperatures of single and binary compounds. Jha et al. [75] predicted polymer glass transition temperatures. Mauro et al. [125] predicted elastic moduli and compressive stress of glass. ...
1
2015
... Machine learning should be viewed as the sum of the organization of the initial datasets, the descriptor creation and algorithm learning steps, and the necessary subsequent steps for targeted new data entry. Ultimately, the expert recommendation system can be improved continuously and adaptively [43]. Material data are made up of datasets, and dataset uncertainties also have a positive impact on machine learning model predictions [75]. Therefore, Bhat et al. [76] established a set of general rules for describing material data that will aid machine learning and decision-making. Material performance can be predicted through machine learning. In other words, the material is first “fingerprinted” (also called generating a descriptor or a feature vector), followed by a mapping between the descriptor and the property of interest, and finally performance prediction [31,43]. No matter what kind of material is studied, the condition of machine learning is the existence of past data. In machine learning algorithms, the input is generally a component or process parameter of the material, and the output is generally the property or properties of interest [77]. Fig. 1 shows the specific process of machine learning [43]. In Fig. 1, N and M are respectively the number of training examples and the number of fingerprint components. ...
4
2015
... Machine learning should be viewed as the sum of the organization of the initial datasets, the descriptor creation and algorithm learning steps, and the necessary subsequent steps for targeted new data entry. Ultimately, the expert recommendation system can be improved continuously and adaptively [43]. Material data are made up of datasets, and dataset uncertainties also have a positive impact on machine learning model predictions [75]. Therefore, Bhat et al. [76] established a set of general rules for describing material data that will aid machine learning and decision-making. Material performance can be predicted through machine learning. In other words, the material is first “fingerprinted” (also called generating a descriptor or a feature vector), followed by a mapping between the descriptor and the property of interest, and finally performance prediction [31,43]. No matter what kind of material is studied, the condition of machine learning is the existence of past data. In machine learning algorithms, the input is generally a component or process parameter of the material, and the output is generally the property or properties of interest [77]. Fig. 1 shows the specific process of machine learning [43]. In Fig. 1, N and M are respectively the number of training examples and the number of fingerprint components. ...
... Recently, machine learning algorithms and statistical inference [90] have often been used in combination. They are mainly used to predict various properties of material systems effectively and accurately [91]. In the future, they may build material knowledge bases through machine learning and statistical inference [92]. In addition, machine learning methods can be combined with various intelligent algorithms, such as genetic algorithms (GA) [77], which are mainly used to optimize the parameters of machine learning models. Tzuc et al. [93] developed a genetic programming model for optimizing zeolite materials adsorption. Furthermore, GA can explore large and complex search spaces very efficiently [94]. When the amount of data is limited, data mining [95] methods can be used to determine the significant correlations between the ab initio energies of different structures in different materials [2,21,29]. Recently, the method of material data mining has developed rapidly [13] and has produced an open-source toolkit for materials data mining called Matminer [96]. Lu et al. [97] have used data mining to design layered double hydroxides with the required specific surface area. ...
... ANN has been widely used in many fields of research [113]; this may have been due to ANN attempts to model the functioning of human brains [114]. For example, at present, ANN has become a popular tool for Al-Si alloy performance prediction [[115], [116], [117]]. ANN has been developed to predict the porosity percentage of Al-Si casting alloys and has been used to correlate chemical composition and cooling rate with porosity [118,119]. An ANN has accurately predicted the corrosion resistance of Al-Si-Mg-based metal matrix composites reinforced with SiC particles, with the average square of the Pearson product-moment correlation coefficient (R2), the maximum mean square error, and the minimum root mean squared deviation calculated as 0.9904, 0.00002476, and 0.00157480, respectively. It can be seen that the experimental results are highly consistent with the ANN results [120]. ANN was used to predict the mechanical properties of A356, including yield stress, ultimate tensile strength, maximum force, and elongation percentage; the predictions of the ANN model were found to be in good agreement with experimental data [121,122]. In addition, ANN models have been used to investigate the role of composition and processing parameters in the mechanical properties of microalloyed pipeline steel and to design steel with improved performance with regard to strength, impact toughness, and ductility. Then the models were used as objective functions for multi-objective genetic algorithms to evolve tradeoffs among the conflicting objectives of improved strength, better ductility, and higher impact toughness. Moreover, the Pareto optimal solutions were successfully analyzed to study the role of various parameters in designing pipeline steel with such improved performance [77]. Fig. 4 shows the specific flow chart. ...
... Flow chart of the computational methodology adopted for designing pipeline steel [77].
Mannodi-Kanakkithodi et al. [123] used a combination of statistical learning and genetic algorithms to predict polymer dielectrics. Within the dataset, the model can not only determine the corresponding properties of any polymer, but can also actively find the specific polymerization that suits its requirements. However, because the model had only seven of the necessary pieces of building-block information, the information guide in the model had certain limitations. ...
1
2015
... As shown in Fig. 2, machine learning algorithms commonly used in materials science can be divided into four categories: probability estimation [20], regression, clustering, and classification [78]. The main machine learning methods for material performance prediction are regression, clustering, and classification algorithms [20]. Examples include kernel ridge regression [79], artificial neural networks (ANN) [[80], [81], [82]], SVM [83], Gaussian process regression [84], feature learning, clustering, matrix factorization, and constraint reasoning [85], Adaboost [86], and decision trees [87]. Machine learning can also be used to discover new materials, for example, using probability estimation algorithms [88] or principal component analysis [2,89]. ...
2
2015
... As shown in Fig. 2, machine learning algorithms commonly used in materials science can be divided into four categories: probability estimation [20], regression, clustering, and classification [78]. The main machine learning methods for material performance prediction are regression, clustering, and classification algorithms [20]. Examples include kernel ridge regression [79], artificial neural networks (ANN) [[80], [81], [82]], SVM [83], Gaussian process regression [84], feature learning, clustering, matrix factorization, and constraint reasoning [85], Adaboost [86], and decision trees [87]. Machine learning can also be used to discover new materials, for example, using probability estimation algorithms [88] or principal component analysis [2,89]. ...
... Predicting and characterizing the crystal structure of materials is a key issue in materials research and development [2,101,102]. However, the accuracy of the prediction depends largely on how the crystals are represented [103]. Faber et al. [79] introduced and evaluated a set of feature vector representations of crystal structures for machine learning models of formation energies of solids. Faber et al. [104] developed all possible elpasolite crystals using a machine learning model. Data mining was used to establish the structural design rules of crystal chemistry. Combined with first-principles calculations, statistical inference can be used as a tool to accelerate significantly the prediction of unknown crystal structures [105]. Machine learning models can quickly predict the properties of crystalline materials [8]. Furthermore, a machine learning model can also predict point defect properties of crystal structures, which was the first application of machine learning in this field [106]. ...
1
2013
... As shown in Fig. 2, machine learning algorithms commonly used in materials science can be divided into four categories: probability estimation [20], regression, clustering, and classification [78]. The main machine learning methods for material performance prediction are regression, clustering, and classification algorithms [20]. Examples include kernel ridge regression [79], artificial neural networks (ANN) [[80], [81], [82]], SVM [83], Gaussian process regression [84], feature learning, clustering, matrix factorization, and constraint reasoning [85], Adaboost [86], and decision trees [87]. Machine learning can also be used to discover new materials, for example, using probability estimation algorithms [88] or principal component analysis [2,89]. ...
1
2013
... As shown in Fig. 2, machine learning algorithms commonly used in materials science can be divided into four categories: probability estimation [20], regression, clustering, and classification [78]. The main machine learning methods for material performance prediction are regression, clustering, and classification algorithms [20]. Examples include kernel ridge regression [79], artificial neural networks (ANN) [[80], [81], [82]], SVM [83], Gaussian process regression [84], feature learning, clustering, matrix factorization, and constraint reasoning [85], Adaboost [86], and decision trees [87]. Machine learning can also be used to discover new materials, for example, using probability estimation algorithms [88] or principal component analysis [2,89]. ...
1
2017
... As shown in Fig. 2, machine learning algorithms commonly used in materials science can be divided into four categories: probability estimation [20], regression, clustering, and classification [78]. The main machine learning methods for material performance prediction are regression, clustering, and classification algorithms [20]. Examples include kernel ridge regression [79], artificial neural networks (ANN) [[80], [81], [82]], SVM [83], Gaussian process regression [84], feature learning, clustering, matrix factorization, and constraint reasoning [85], Adaboost [86], and decision trees [87]. Machine learning can also be used to discover new materials, for example, using probability estimation algorithms [88] or principal component analysis [2,89]. ...
2
2016
... As shown in Fig. 2, machine learning algorithms commonly used in materials science can be divided into four categories: probability estimation [20], regression, clustering, and classification [78]. The main machine learning methods for material performance prediction are regression, clustering, and classification algorithms [20]. Examples include kernel ridge regression [79], artificial neural networks (ANN) [[80], [81], [82]], SVM [83], Gaussian process regression [84], feature learning, clustering, matrix factorization, and constraint reasoning [85], Adaboost [86], and decision trees [87]. Machine learning can also be used to discover new materials, for example, using probability estimation algorithms [88] or principal component analysis [2,89]. ...
... To accelerate the design of new materials, recent studies have screened out many types of promising candidate materials [51], including perovskite compounds [83,142], metal-organic frameworks [143,144], catalysts [145,146], light-emitting molecules [147], and thermoelectric materials [[148], [149], [150], [151]]. A large-scale database based on quantum mechanical calculation of known materials, using machine learning to calculate material screening, is a combination screening process for new materials in unconstrained component space, which can be used for an extensive range of critical material discoveries [87]. In addition, interface structure and energy can be also predicted through virtual screening [152]. ...
1
2010
... As shown in Fig. 2, machine learning algorithms commonly used in materials science can be divided into four categories: probability estimation [20], regression, clustering, and classification [78]. The main machine learning methods for material performance prediction are regression, clustering, and classification algorithms [20]. Examples include kernel ridge regression [79], artificial neural networks (ANN) [[80], [81], [82]], SVM [83], Gaussian process regression [84], feature learning, clustering, matrix factorization, and constraint reasoning [85], Adaboost [86], and decision trees [87]. Machine learning can also be used to discover new materials, for example, using probability estimation algorithms [88] or principal component analysis [2,89]. ...
2
2016
... As shown in Fig. 2, machine learning algorithms commonly used in materials science can be divided into four categories: probability estimation [20], regression, clustering, and classification [78]. The main machine learning methods for material performance prediction are regression, clustering, and classification algorithms [20]. Examples include kernel ridge regression [79], artificial neural networks (ANN) [[80], [81], [82]], SVM [83], Gaussian process regression [84], feature learning, clustering, matrix factorization, and constraint reasoning [85], Adaboost [86], and decision trees [87]. Machine learning can also be used to discover new materials, for example, using probability estimation algorithms [88] or principal component analysis [2,89]. ...
... High-throughput experimentation studies have resulted in large, rapidly growing volumes of structured data, making expert manual analysis impractical. Therefore, the materials community has turned to fast data analysis techniques for machine learning to convert large amounts of structured data into phase diagrams. Although machine learning promises to provide high-throughput phase diagram determination, its success depends on several factors—prior knowledge, data preprocessing, data representation, similarity or dissimilarity measures, and model choice [85]. Rajan et al. [98] selected principal component analysis, partial least-squares regression, and correlation function expansion to perform data mining on compound semiconductor property phase diagrams. Artrith et al. [99] used a combination ANN and GA algorithm to accelerate sampling of amorphous and disordered materials and constructed a first-principles phase diagram of an amorphous LiSi alloy. This reference showed that using this combined machine learning model can accelerate first-principles sampling of complex structure spaces, proved that this combined model is more efficient than the individual ANN model, and determined that sampling was successful in determining low-energy amorphous structures. Spellings et al. [100] used machine learning to discover interesting regions of the parameter space in colloid self-assembly and created descriptors, then located interesting regions of complex phase diagrams without a priori information, and finally used knowledge of available structures to generate a phase diagram automatically. Fig. 3 shows the phase diagram generated by the Gaussian mixture model. ...
1
2015
... As shown in Fig. 2, machine learning algorithms commonly used in materials science can be divided into four categories: probability estimation [20], regression, clustering, and classification [78]. The main machine learning methods for material performance prediction are regression, clustering, and classification algorithms [20]. Examples include kernel ridge regression [79], artificial neural networks (ANN) [[80], [81], [82]], SVM [83], Gaussian process regression [84], feature learning, clustering, matrix factorization, and constraint reasoning [85], Adaboost [86], and decision trees [87]. Machine learning can also be used to discover new materials, for example, using probability estimation algorithms [88] or principal component analysis [2,89]. ...
2
2014
... As shown in Fig. 2, machine learning algorithms commonly used in materials science can be divided into four categories: probability estimation [20], regression, clustering, and classification [78]. The main machine learning methods for material performance prediction are regression, clustering, and classification algorithms [20]. Examples include kernel ridge regression [79], artificial neural networks (ANN) [[80], [81], [82]], SVM [83], Gaussian process regression [84], feature learning, clustering, matrix factorization, and constraint reasoning [85], Adaboost [86], and decision trees [87]. Machine learning can also be used to discover new materials, for example, using probability estimation algorithms [88] or principal component analysis [2,89]. ...
... To accelerate the design of new materials, recent studies have screened out many types of promising candidate materials [51], including perovskite compounds [83,142], metal-organic frameworks [143,144], catalysts [145,146], light-emitting molecules [147], and thermoelectric materials [[148], [149], [150], [151]]. A large-scale database based on quantum mechanical calculation of known materials, using machine learning to calculate material screening, is a combination screening process for new materials in unconstrained component space, which can be used for an extensive range of critical material discoveries [87]. In addition, interface structure and energy can be also predicted through virtual screening [152]. ...
1
2019
... As shown in Fig. 2, machine learning algorithms commonly used in materials science can be divided into four categories: probability estimation [20], regression, clustering, and classification [78]. The main machine learning methods for material performance prediction are regression, clustering, and classification algorithms [20]. Examples include kernel ridge regression [79], artificial neural networks (ANN) [[80], [81], [82]], SVM [83], Gaussian process regression [84], feature learning, clustering, matrix factorization, and constraint reasoning [85], Adaboost [86], and decision trees [87]. Machine learning can also be used to discover new materials, for example, using probability estimation algorithms [88] or principal component analysis [2,89]. ...
1
2015
... As shown in Fig. 2, machine learning algorithms commonly used in materials science can be divided into four categories: probability estimation [20], regression, clustering, and classification [78]. The main machine learning methods for material performance prediction are regression, clustering, and classification algorithms [20]. Examples include kernel ridge regression [79], artificial neural networks (ANN) [[80], [81], [82]], SVM [83], Gaussian process regression [84], feature learning, clustering, matrix factorization, and constraint reasoning [85], Adaboost [86], and decision trees [87]. Machine learning can also be used to discover new materials, for example, using probability estimation algorithms [88] or principal component analysis [2,89]. ...
1
2017
... Recently, machine learning algorithms and statistical inference [90] have often been used in combination. They are mainly used to predict various properties of material systems effectively and accurately [91]. In the future, they may build material knowledge bases through machine learning and statistical inference [92]. In addition, machine learning methods can be combined with various intelligent algorithms, such as genetic algorithms (GA) [77], which are mainly used to optimize the parameters of machine learning models. Tzuc et al. [93] developed a genetic programming model for optimizing zeolite materials adsorption. Furthermore, GA can explore large and complex search spaces very efficiently [94]. When the amount of data is limited, data mining [95] methods can be used to determine the significant correlations between the ab initio energies of different structures in different materials [2,21,29]. Recently, the method of material data mining has developed rapidly [13] and has produced an open-source toolkit for materials data mining called Matminer [96]. Lu et al. [97] have used data mining to design layered double hydroxides with the required specific surface area. ...
2
2013
... Recently, machine learning algorithms and statistical inference [90] have often been used in combination. They are mainly used to predict various properties of material systems effectively and accurately [91]. In the future, they may build material knowledge bases through machine learning and statistical inference [92]. In addition, machine learning methods can be combined with various intelligent algorithms, such as genetic algorithms (GA) [77], which are mainly used to optimize the parameters of machine learning models. Tzuc et al. [93] developed a genetic programming model for optimizing zeolite materials adsorption. Furthermore, GA can explore large and complex search spaces very efficiently [94]. When the amount of data is limited, data mining [95] methods can be used to determine the significant correlations between the ab initio energies of different structures in different materials [2,21,29]. Recently, the method of material data mining has developed rapidly [13] and has produced an open-source toolkit for materials data mining called Matminer [96]. Lu et al. [97] have used data mining to design layered double hydroxides with the required specific surface area. ...
... If people can effectively learn from available knowledge and data, the material discovery process can be significantly accelerated and simplified. Using a one-dimensional chain system family, Pilania et al. [91] proposed a general form that can discover decision rules and establish a mapping between attributes that are easily accessible to the system and their properties. The results show that fingerprints based on either chemical structure (compositional and configurational information) or electronic charge density distribution can be used for ultra-fast, but accurate property predictions. Harnessing such learning paradigms extends recent efforts to explore systematically huge chemical spaces and can significantly accelerate discovery of new application-specific materials. Curtarolo et al. [135] introduced new material fingerprint descriptors that produce a material mapping network: nodes represent compounds, and connections represent similarities. The mapping network can identify compounds with different physical and chemical properties. Recently, the t-student stochastic neighbor embedding algorithm was used for the first time to extract feature spaces from band-structure ab initio calculations. This algorithm can design different spaces and map them to lower dimensions, enabling simultaneous analysis and exploration of previously unknown band structures for thousands of materials. The more information is available in the material database, the more space can be explored [136]. Ramprasad et al. [137] have studied a series of motif-based topological fingerprints that can represent the major classes of crystals and molecules by numbers. By using a learning algorithm, these fingerprints can be mapped to various properties of crystals and molecules, thereby accelerating property prediction capabilities. Their paper proves the process by simultaneously optimizing two properties, the gap between the highest occupied and lowest unoccupied molecular orbitals EHL and the isotropic polarizability α. The results are shown in Fig. 6 and Table 1, which show that molecules with the required values of EHL and α were obtained. ...
1
2012
... Recently, machine learning algorithms and statistical inference [90] have often been used in combination. They are mainly used to predict various properties of material systems effectively and accurately [91]. In the future, they may build material knowledge bases through machine learning and statistical inference [92]. In addition, machine learning methods can be combined with various intelligent algorithms, such as genetic algorithms (GA) [77], which are mainly used to optimize the parameters of machine learning models. Tzuc et al. [93] developed a genetic programming model for optimizing zeolite materials adsorption. Furthermore, GA can explore large and complex search spaces very efficiently [94]. When the amount of data is limited, data mining [95] methods can be used to determine the significant correlations between the ab initio energies of different structures in different materials [2,21,29]. Recently, the method of material data mining has developed rapidly [13] and has produced an open-source toolkit for materials data mining called Matminer [96]. Lu et al. [97] have used data mining to design layered double hydroxides with the required specific surface area. ...
1
2018
... Recently, machine learning algorithms and statistical inference [90] have often been used in combination. They are mainly used to predict various properties of material systems effectively and accurately [91]. In the future, they may build material knowledge bases through machine learning and statistical inference [92]. In addition, machine learning methods can be combined with various intelligent algorithms, such as genetic algorithms (GA) [77], which are mainly used to optimize the parameters of machine learning models. Tzuc et al. [93] developed a genetic programming model for optimizing zeolite materials adsorption. Furthermore, GA can explore large and complex search spaces very efficiently [94]. When the amount of data is limited, data mining [95] methods can be used to determine the significant correlations between the ab initio energies of different structures in different materials [2,21,29]. Recently, the method of material data mining has developed rapidly [13] and has produced an open-source toolkit for materials data mining called Matminer [96]. Lu et al. [97] have used data mining to design layered double hydroxides with the required specific surface area. ...
1
2016
... Recently, machine learning algorithms and statistical inference [90] have often been used in combination. They are mainly used to predict various properties of material systems effectively and accurately [91]. In the future, they may build material knowledge bases through machine learning and statistical inference [92]. In addition, machine learning methods can be combined with various intelligent algorithms, such as genetic algorithms (GA) [77], which are mainly used to optimize the parameters of machine learning models. Tzuc et al. [93] developed a genetic programming model for optimizing zeolite materials adsorption. Furthermore, GA can explore large and complex search spaces very efficiently [94]. When the amount of data is limited, data mining [95] methods can be used to determine the significant correlations between the ab initio energies of different structures in different materials [2,21,29]. Recently, the method of material data mining has developed rapidly [13] and has produced an open-source toolkit for materials data mining called Matminer [96]. Lu et al. [97] have used data mining to design layered double hydroxides with the required specific surface area. ...
1
2016
... Recently, machine learning algorithms and statistical inference [90] have often been used in combination. They are mainly used to predict various properties of material systems effectively and accurately [91]. In the future, they may build material knowledge bases through machine learning and statistical inference [92]. In addition, machine learning methods can be combined with various intelligent algorithms, such as genetic algorithms (GA) [77], which are mainly used to optimize the parameters of machine learning models. Tzuc et al. [93] developed a genetic programming model for optimizing zeolite materials adsorption. Furthermore, GA can explore large and complex search spaces very efficiently [94]. When the amount of data is limited, data mining [95] methods can be used to determine the significant correlations between the ab initio energies of different structures in different materials [2,21,29]. Recently, the method of material data mining has developed rapidly [13] and has produced an open-source toolkit for materials data mining called Matminer [96]. Lu et al. [97] have used data mining to design layered double hydroxides with the required specific surface area. ...
1
2018
... Recently, machine learning algorithms and statistical inference [90] have often been used in combination. They are mainly used to predict various properties of material systems effectively and accurately [91]. In the future, they may build material knowledge bases through machine learning and statistical inference [92]. In addition, machine learning methods can be combined with various intelligent algorithms, such as genetic algorithms (GA) [77], which are mainly used to optimize the parameters of machine learning models. Tzuc et al. [93] developed a genetic programming model for optimizing zeolite materials adsorption. Furthermore, GA can explore large and complex search spaces very efficiently [94]. When the amount of data is limited, data mining [95] methods can be used to determine the significant correlations between the ab initio energies of different structures in different materials [2,21,29]. Recently, the method of material data mining has developed rapidly [13] and has produced an open-source toolkit for materials data mining called Matminer [96]. Lu et al. [97] have used data mining to design layered double hydroxides with the required specific surface area. ...
1
2017
... Recently, machine learning algorithms and statistical inference [90] have often been used in combination. They are mainly used to predict various properties of material systems effectively and accurately [91]. In the future, they may build material knowledge bases through machine learning and statistical inference [92]. In addition, machine learning methods can be combined with various intelligent algorithms, such as genetic algorithms (GA) [77], which are mainly used to optimize the parameters of machine learning models. Tzuc et al. [93] developed a genetic programming model for optimizing zeolite materials adsorption. Furthermore, GA can explore large and complex search spaces very efficiently [94]. When the amount of data is limited, data mining [95] methods can be used to determine the significant correlations between the ab initio energies of different structures in different materials [2,21,29]. Recently, the method of material data mining has developed rapidly [13] and has produced an open-source toolkit for materials data mining called Matminer [96]. Lu et al. [97] have used data mining to design layered double hydroxides with the required specific surface area. ...
1
2013
... High-throughput experimentation studies have resulted in large, rapidly growing volumes of structured data, making expert manual analysis impractical. Therefore, the materials community has turned to fast data analysis techniques for machine learning to convert large amounts of structured data into phase diagrams. Although machine learning promises to provide high-throughput phase diagram determination, its success depends on several factors—prior knowledge, data preprocessing, data representation, similarity or dissimilarity measures, and model choice [85]. Rajan et al. [98] selected principal component analysis, partial least-squares regression, and correlation function expansion to perform data mining on compound semiconductor property phase diagrams. Artrith et al. [99] used a combination ANN and GA algorithm to accelerate sampling of amorphous and disordered materials and constructed a first-principles phase diagram of an amorphous LiSi alloy. This reference showed that using this combined machine learning model can accelerate first-principles sampling of complex structure spaces, proved that this combined model is more efficient than the individual ANN model, and determined that sampling was successful in determining low-energy amorphous structures. Spellings et al. [100] used machine learning to discover interesting regions of the parameter space in colloid self-assembly and created descriptors, then located interesting regions of complex phase diagrams without a priori information, and finally used knowledge of available structures to generate a phase diagram automatically. Fig. 3 shows the phase diagram generated by the Gaussian mixture model. ...
1
2018
... High-throughput experimentation studies have resulted in large, rapidly growing volumes of structured data, making expert manual analysis impractical. Therefore, the materials community has turned to fast data analysis techniques for machine learning to convert large amounts of structured data into phase diagrams. Although machine learning promises to provide high-throughput phase diagram determination, its success depends on several factors—prior knowledge, data preprocessing, data representation, similarity or dissimilarity measures, and model choice [85]. Rajan et al. [98] selected principal component analysis, partial least-squares regression, and correlation function expansion to perform data mining on compound semiconductor property phase diagrams. Artrith et al. [99] used a combination ANN and GA algorithm to accelerate sampling of amorphous and disordered materials and constructed a first-principles phase diagram of an amorphous LiSi alloy. This reference showed that using this combined machine learning model can accelerate first-principles sampling of complex structure spaces, proved that this combined model is more efficient than the individual ANN model, and determined that sampling was successful in determining low-energy amorphous structures. Spellings et al. [100] used machine learning to discover interesting regions of the parameter space in colloid self-assembly and created descriptors, then located interesting regions of complex phase diagrams without a priori information, and finally used knowledge of available structures to generate a phase diagram automatically. Fig. 3 shows the phase diagram generated by the Gaussian mixture model. ...
2
2018
... High-throughput experimentation studies have resulted in large, rapidly growing volumes of structured data, making expert manual analysis impractical. Therefore, the materials community has turned to fast data analysis techniques for machine learning to convert large amounts of structured data into phase diagrams. Although machine learning promises to provide high-throughput phase diagram determination, its success depends on several factors—prior knowledge, data preprocessing, data representation, similarity or dissimilarity measures, and model choice [85]. Rajan et al. [98] selected principal component analysis, partial least-squares regression, and correlation function expansion to perform data mining on compound semiconductor property phase diagrams. Artrith et al. [99] used a combination ANN and GA algorithm to accelerate sampling of amorphous and disordered materials and constructed a first-principles phase diagram of an amorphous LiSi alloy. This reference showed that using this combined machine learning model can accelerate first-principles sampling of complex structure spaces, proved that this combined model is more efficient than the individual ANN model, and determined that sampling was successful in determining low-energy amorphous structures. Spellings et al. [100] used machine learning to discover interesting regions of the parameter space in colloid self-assembly and created descriptors, then located interesting regions of complex phase diagrams without a priori information, and finally used knowledge of available structures to generate a phase diagram automatically. Fig. 3 shows the phase diagram generated by the Gaussian mixture model. ...
... (a) Shannon entropy (blue line) of the quasicrystal data set as GMM components are successively merged from 15 clusters to one cluster. Merged cluster counts corresponding to (b-d) are indicated by black points. (b-d) Phase diagrams generated by taking the most common predicted cluster type for each parameter point, indicated by the black points in (a). For each selected cluster count, dark gray regions show a poor preference for any single structure among the samples for those parameters. Each type of system, as identified by the GMM is assigned a different color, but this unsupervised algorithm clusters the distinct structures that it finds rather than labeling a previously identified set of known structures. Phase boundaries generated by manual analysis are included for reference as black lines [100]. ...
2
2016
... Predicting and characterizing the crystal structure of materials is a key issue in materials research and development [2,101,102]. However, the accuracy of the prediction depends largely on how the crystals are represented [103]. Faber et al. [79] introduced and evaluated a set of feature vector representations of crystal structures for machine learning models of formation energies of solids. Faber et al. [104] developed all possible elpasolite crystals using a machine learning model. Data mining was used to establish the structural design rules of crystal chemistry. Combined with first-principles calculations, statistical inference can be used as a tool to accelerate significantly the prediction of unknown crystal structures [105]. Machine learning models can quickly predict the properties of crystalline materials [8]. Furthermore, a machine learning model can also predict point defect properties of crystal structures, which was the first application of machine learning in this field [106]. ...
... An adaptive design strategy, tightly coupled with experiments, can accelerate the discovery process by sequentially identifying the next experiments or calculations to effectively navigate the sophisticated search space. The strategy uses inference and global optimization to perform the tradeoff between exploitation and exploration of the search space. Oliynyk et al. [138] used real experimental data, selected a suitable machine learning model, and predicted the model through experiments. Finally, many kinds of intermetallic compounds were discovered. The most crucial finding was RhCd, the first new binary AB compound to be found with a CsCl-type structure in over 15 years. Xue et al. [139] demonstrated this by finding very low thermal hysteresis (DT) NiTi-based shape memory alloys, with Ti50.0Ni46.7Cu0.8Fe2.3Pd0.2 possessing the smallest DT (1.84░K). Wolverton et al. [101] used a general machine learning framework to predict the properties of various materials and discovered a metallic glass alloy. Mauro et al. [125] developed new damage-resistant glasses. Ren et al. [140] found metallic glasses through iterations of machine learning. Yuan et al. [141] used a combination of machine learning and optimization methods to accelerate the discovery of new Pb-free BaTiO3-based piezoelectrics with large electro strains. ...
1
2015
... Predicting and characterizing the crystal structure of materials is a key issue in materials research and development [2,101,102]. However, the accuracy of the prediction depends largely on how the crystals are represented [103]. Faber et al. [79] introduced and evaluated a set of feature vector representations of crystal structures for machine learning models of formation energies of solids. Faber et al. [104] developed all possible elpasolite crystals using a machine learning model. Data mining was used to establish the structural design rules of crystal chemistry. Combined with first-principles calculations, statistical inference can be used as a tool to accelerate significantly the prediction of unknown crystal structures [105]. Machine learning models can quickly predict the properties of crystalline materials [8]. Furthermore, a machine learning model can also predict point defect properties of crystal structures, which was the first application of machine learning in this field [106]. ...
1
2014
... Predicting and characterizing the crystal structure of materials is a key issue in materials research and development [2,101,102]. However, the accuracy of the prediction depends largely on how the crystals are represented [103]. Faber et al. [79] introduced and evaluated a set of feature vector representations of crystal structures for machine learning models of formation energies of solids. Faber et al. [104] developed all possible elpasolite crystals using a machine learning model. Data mining was used to establish the structural design rules of crystal chemistry. Combined with first-principles calculations, statistical inference can be used as a tool to accelerate significantly the prediction of unknown crystal structures [105]. Machine learning models can quickly predict the properties of crystalline materials [8]. Furthermore, a machine learning model can also predict point defect properties of crystal structures, which was the first application of machine learning in this field [106]. ...
1
2016
... Predicting and characterizing the crystal structure of materials is a key issue in materials research and development [2,101,102]. However, the accuracy of the prediction depends largely on how the crystals are represented [103]. Faber et al. [79] introduced and evaluated a set of feature vector representations of crystal structures for machine learning models of formation energies of solids. Faber et al. [104] developed all possible elpasolite crystals using a machine learning model. Data mining was used to establish the structural design rules of crystal chemistry. Combined with first-principles calculations, statistical inference can be used as a tool to accelerate significantly the prediction of unknown crystal structures [105]. Machine learning models can quickly predict the properties of crystalline materials [8]. Furthermore, a machine learning model can also predict point defect properties of crystal structures, which was the first application of machine learning in this field [106]. ...
1
2012
... Predicting and characterizing the crystal structure of materials is a key issue in materials research and development [2,101,102]. However, the accuracy of the prediction depends largely on how the crystals are represented [103]. Faber et al. [79] introduced and evaluated a set of feature vector representations of crystal structures for machine learning models of formation energies of solids. Faber et al. [104] developed all possible elpasolite crystals using a machine learning model. Data mining was used to establish the structural design rules of crystal chemistry. Combined with first-principles calculations, statistical inference can be used as a tool to accelerate significantly the prediction of unknown crystal structures [105]. Machine learning models can quickly predict the properties of crystalline materials [8]. Furthermore, a machine learning model can also predict point defect properties of crystal structures, which was the first application of machine learning in this field [106]. ...
1
2016
... Predicting and characterizing the crystal structure of materials is a key issue in materials research and development [2,101,102]. However, the accuracy of the prediction depends largely on how the crystals are represented [103]. Faber et al. [79] introduced and evaluated a set of feature vector representations of crystal structures for machine learning models of formation energies of solids. Faber et al. [104] developed all possible elpasolite crystals using a machine learning model. Data mining was used to establish the structural design rules of crystal chemistry. Combined with first-principles calculations, statistical inference can be used as a tool to accelerate significantly the prediction of unknown crystal structures [105]. Machine learning models can quickly predict the properties of crystalline materials [8]. Furthermore, a machine learning model can also predict point defect properties of crystal structures, which was the first application of machine learning in this field [106]. ...
1
2009
... Finding new materials with targeted properties has traditionally been guided by intuition plus trial and error. At present, with the increasing amount of data available, using machine learning to accelerate the prediction of material properties and then to discover these new materials has become a new method. Research on material properties has focused on the relationship between the properties of materials and their microstructure [107,108]. Shi et al. [20] pointed out that machine learning applications to material property prediction can be divided into two categories: macroscopic performance prediction and microscopic property prediction. ...
1
2008
... Finding new materials with targeted properties has traditionally been guided by intuition plus trial and error. At present, with the increasing amount of data available, using machine learning to accelerate the prediction of material properties and then to discover these new materials has become a new method. Research on material properties has focused on the relationship between the properties of materials and their microstructure [107,108]. Shi et al. [20] pointed out that machine learning applications to material property prediction can be divided into two categories: macroscopic performance prediction and microscopic property prediction. ...
1
2007
... An ANN uses massively parallel distributed processors based on data, which tend to store experiential knowledge and make it available for use [109]. ANN theory learns from previously obtained data, which are called training sets, and then checks the system accuracy using test data [110]. ANN is very powerful in solving nonlinear problems [111]. However, artificial neural networks have the drawback of over-fitting, but Bayesian regularization can solve this problem [112]. ...
1
2004
... An ANN uses massively parallel distributed processors based on data, which tend to store experiential knowledge and make it available for use [109]. ANN theory learns from previously obtained data, which are called training sets, and then checks the system accuracy using test data [110]. ANN is very powerful in solving nonlinear problems [111]. However, artificial neural networks have the drawback of over-fitting, but Bayesian regularization can solve this problem [112]. ...
1
2014
... An ANN uses massively parallel distributed processors based on data, which tend to store experiential knowledge and make it available for use [109]. ANN theory learns from previously obtained data, which are called training sets, and then checks the system accuracy using test data [110]. ANN is very powerful in solving nonlinear problems [111]. However, artificial neural networks have the drawback of over-fitting, but Bayesian regularization can solve this problem [112]. ...
1
2018
... An ANN uses massively parallel distributed processors based on data, which tend to store experiential knowledge and make it available for use [109]. ANN theory learns from previously obtained data, which are called training sets, and then checks the system accuracy using test data [110]. ANN is very powerful in solving nonlinear problems [111]. However, artificial neural networks have the drawback of over-fitting, but Bayesian regularization can solve this problem [112]. ...
1
2009
... ANN has been widely used in many fields of research [113]; this may have been due to ANN attempts to model the functioning of human brains [114]. For example, at present, ANN has become a popular tool for Al-Si alloy performance prediction [[115], [116], [117]]. ANN has been developed to predict the porosity percentage of Al-Si casting alloys and has been used to correlate chemical composition and cooling rate with porosity [118,119]. An ANN has accurately predicted the corrosion resistance of Al-Si-Mg-based metal matrix composites reinforced with SiC particles, with the average square of the Pearson product-moment correlation coefficient (R2), the maximum mean square error, and the minimum root mean squared deviation calculated as 0.9904, 0.00002476, and 0.00157480, respectively. It can be seen that the experimental results are highly consistent with the ANN results [120]. ANN was used to predict the mechanical properties of A356, including yield stress, ultimate tensile strength, maximum force, and elongation percentage; the predictions of the ANN model were found to be in good agreement with experimental data [121,122]. In addition, ANN models have been used to investigate the role of composition and processing parameters in the mechanical properties of microalloyed pipeline steel and to design steel with improved performance with regard to strength, impact toughness, and ductility. Then the models were used as objective functions for multi-objective genetic algorithms to evolve tradeoffs among the conflicting objectives of improved strength, better ductility, and higher impact toughness. Moreover, the Pareto optimal solutions were successfully analyzed to study the role of various parameters in designing pipeline steel with such improved performance [77]. Fig. 4 shows the specific flow chart. ...
1
2009
... ANN has been widely used in many fields of research [113]; this may have been due to ANN attempts to model the functioning of human brains [114]. For example, at present, ANN has become a popular tool for Al-Si alloy performance prediction [[115], [116], [117]]. ANN has been developed to predict the porosity percentage of Al-Si casting alloys and has been used to correlate chemical composition and cooling rate with porosity [118,119]. An ANN has accurately predicted the corrosion resistance of Al-Si-Mg-based metal matrix composites reinforced with SiC particles, with the average square of the Pearson product-moment correlation coefficient (R2), the maximum mean square error, and the minimum root mean squared deviation calculated as 0.9904, 0.00002476, and 0.00157480, respectively. It can be seen that the experimental results are highly consistent with the ANN results [120]. ANN was used to predict the mechanical properties of A356, including yield stress, ultimate tensile strength, maximum force, and elongation percentage; the predictions of the ANN model were found to be in good agreement with experimental data [121,122]. In addition, ANN models have been used to investigate the role of composition and processing parameters in the mechanical properties of microalloyed pipeline steel and to design steel with improved performance with regard to strength, impact toughness, and ductility. Then the models were used as objective functions for multi-objective genetic algorithms to evolve tradeoffs among the conflicting objectives of improved strength, better ductility, and higher impact toughness. Moreover, the Pareto optimal solutions were successfully analyzed to study the role of various parameters in designing pipeline steel with such improved performance [77]. Fig. 4 shows the specific flow chart. ...
1
2011
... ANN has been widely used in many fields of research [113]; this may have been due to ANN attempts to model the functioning of human brains [114]. For example, at present, ANN has become a popular tool for Al-Si alloy performance prediction [[115], [116], [117]]. ANN has been developed to predict the porosity percentage of Al-Si casting alloys and has been used to correlate chemical composition and cooling rate with porosity [118,119]. An ANN has accurately predicted the corrosion resistance of Al-Si-Mg-based metal matrix composites reinforced with SiC particles, with the average square of the Pearson product-moment correlation coefficient (R2), the maximum mean square error, and the minimum root mean squared deviation calculated as 0.9904, 0.00002476, and 0.00157480, respectively. It can be seen that the experimental results are highly consistent with the ANN results [120]. ANN was used to predict the mechanical properties of A356, including yield stress, ultimate tensile strength, maximum force, and elongation percentage; the predictions of the ANN model were found to be in good agreement with experimental data [121,122]. In addition, ANN models have been used to investigate the role of composition and processing parameters in the mechanical properties of microalloyed pipeline steel and to design steel with improved performance with regard to strength, impact toughness, and ductility. Then the models were used as objective functions for multi-objective genetic algorithms to evolve tradeoffs among the conflicting objectives of improved strength, better ductility, and higher impact toughness. Moreover, the Pareto optimal solutions were successfully analyzed to study the role of various parameters in designing pipeline steel with such improved performance [77]. Fig. 4 shows the specific flow chart. ...
1
1998
... ANN has been widely used in many fields of research [113]; this may have been due to ANN attempts to model the functioning of human brains [114]. For example, at present, ANN has become a popular tool for Al-Si alloy performance prediction [[115], [116], [117]]. ANN has been developed to predict the porosity percentage of Al-Si casting alloys and has been used to correlate chemical composition and cooling rate with porosity [118,119]. An ANN has accurately predicted the corrosion resistance of Al-Si-Mg-based metal matrix composites reinforced with SiC particles, with the average square of the Pearson product-moment correlation coefficient (R2), the maximum mean square error, and the minimum root mean squared deviation calculated as 0.9904, 0.00002476, and 0.00157480, respectively. It can be seen that the experimental results are highly consistent with the ANN results [120]. ANN was used to predict the mechanical properties of A356, including yield stress, ultimate tensile strength, maximum force, and elongation percentage; the predictions of the ANN model were found to be in good agreement with experimental data [121,122]. In addition, ANN models have been used to investigate the role of composition and processing parameters in the mechanical properties of microalloyed pipeline steel and to design steel with improved performance with regard to strength, impact toughness, and ductility. Then the models were used as objective functions for multi-objective genetic algorithms to evolve tradeoffs among the conflicting objectives of improved strength, better ductility, and higher impact toughness. Moreover, the Pareto optimal solutions were successfully analyzed to study the role of various parameters in designing pipeline steel with such improved performance [77]. Fig. 4 shows the specific flow chart. ...
1
2009
... ANN has been widely used in many fields of research [113]; this may have been due to ANN attempts to model the functioning of human brains [114]. For example, at present, ANN has become a popular tool for Al-Si alloy performance prediction [[115], [116], [117]]. ANN has been developed to predict the porosity percentage of Al-Si casting alloys and has been used to correlate chemical composition and cooling rate with porosity [118,119]. An ANN has accurately predicted the corrosion resistance of Al-Si-Mg-based metal matrix composites reinforced with SiC particles, with the average square of the Pearson product-moment correlation coefficient (R2), the maximum mean square error, and the minimum root mean squared deviation calculated as 0.9904, 0.00002476, and 0.00157480, respectively. It can be seen that the experimental results are highly consistent with the ANN results [120]. ANN was used to predict the mechanical properties of A356, including yield stress, ultimate tensile strength, maximum force, and elongation percentage; the predictions of the ANN model were found to be in good agreement with experimental data [121,122]. In addition, ANN models have been used to investigate the role of composition and processing parameters in the mechanical properties of microalloyed pipeline steel and to design steel with improved performance with regard to strength, impact toughness, and ductility. Then the models were used as objective functions for multi-objective genetic algorithms to evolve tradeoffs among the conflicting objectives of improved strength, better ductility, and higher impact toughness. Moreover, the Pareto optimal solutions were successfully analyzed to study the role of various parameters in designing pipeline steel with such improved performance [77]. Fig. 4 shows the specific flow chart. ...
1
2006
... ANN has been widely used in many fields of research [113]; this may have been due to ANN attempts to model the functioning of human brains [114]. For example, at present, ANN has become a popular tool for Al-Si alloy performance prediction [[115], [116], [117]]. ANN has been developed to predict the porosity percentage of Al-Si casting alloys and has been used to correlate chemical composition and cooling rate with porosity [118,119]. An ANN has accurately predicted the corrosion resistance of Al-Si-Mg-based metal matrix composites reinforced with SiC particles, with the average square of the Pearson product-moment correlation coefficient (R2), the maximum mean square error, and the minimum root mean squared deviation calculated as 0.9904, 0.00002476, and 0.00157480, respectively. It can be seen that the experimental results are highly consistent with the ANN results [120]. ANN was used to predict the mechanical properties of A356, including yield stress, ultimate tensile strength, maximum force, and elongation percentage; the predictions of the ANN model were found to be in good agreement with experimental data [121,122]. In addition, ANN models have been used to investigate the role of composition and processing parameters in the mechanical properties of microalloyed pipeline steel and to design steel with improved performance with regard to strength, impact toughness, and ductility. Then the models were used as objective functions for multi-objective genetic algorithms to evolve tradeoffs among the conflicting objectives of improved strength, better ductility, and higher impact toughness. Moreover, the Pareto optimal solutions were successfully analyzed to study the role of various parameters in designing pipeline steel with such improved performance [77]. Fig. 4 shows the specific flow chart. ...
1
2006
... ANN has been widely used in many fields of research [113]; this may have been due to ANN attempts to model the functioning of human brains [114]. For example, at present, ANN has become a popular tool for Al-Si alloy performance prediction [[115], [116], [117]]. ANN has been developed to predict the porosity percentage of Al-Si casting alloys and has been used to correlate chemical composition and cooling rate with porosity [118,119]. An ANN has accurately predicted the corrosion resistance of Al-Si-Mg-based metal matrix composites reinforced with SiC particles, with the average square of the Pearson product-moment correlation coefficient (R2), the maximum mean square error, and the minimum root mean squared deviation calculated as 0.9904, 0.00002476, and 0.00157480, respectively. It can be seen that the experimental results are highly consistent with the ANN results [120]. ANN was used to predict the mechanical properties of A356, including yield stress, ultimate tensile strength, maximum force, and elongation percentage; the predictions of the ANN model were found to be in good agreement with experimental data [121,122]. In addition, ANN models have been used to investigate the role of composition and processing parameters in the mechanical properties of microalloyed pipeline steel and to design steel with improved performance with regard to strength, impact toughness, and ductility. Then the models were used as objective functions for multi-objective genetic algorithms to evolve tradeoffs among the conflicting objectives of improved strength, better ductility, and higher impact toughness. Moreover, the Pareto optimal solutions were successfully analyzed to study the role of various parameters in designing pipeline steel with such improved performance [77]. Fig. 4 shows the specific flow chart. ...
1
2015
... ANN has been widely used in many fields of research [113]; this may have been due to ANN attempts to model the functioning of human brains [114]. For example, at present, ANN has become a popular tool for Al-Si alloy performance prediction [[115], [116], [117]]. ANN has been developed to predict the porosity percentage of Al-Si casting alloys and has been used to correlate chemical composition and cooling rate with porosity [118,119]. An ANN has accurately predicted the corrosion resistance of Al-Si-Mg-based metal matrix composites reinforced with SiC particles, with the average square of the Pearson product-moment correlation coefficient (R2), the maximum mean square error, and the minimum root mean squared deviation calculated as 0.9904, 0.00002476, and 0.00157480, respectively. It can be seen that the experimental results are highly consistent with the ANN results [120]. ANN was used to predict the mechanical properties of A356, including yield stress, ultimate tensile strength, maximum force, and elongation percentage; the predictions of the ANN model were found to be in good agreement with experimental data [121,122]. In addition, ANN models have been used to investigate the role of composition and processing parameters in the mechanical properties of microalloyed pipeline steel and to design steel with improved performance with regard to strength, impact toughness, and ductility. Then the models were used as objective functions for multi-objective genetic algorithms to evolve tradeoffs among the conflicting objectives of improved strength, better ductility, and higher impact toughness. Moreover, the Pareto optimal solutions were successfully analyzed to study the role of various parameters in designing pipeline steel with such improved performance [77]. Fig. 4 shows the specific flow chart. ...
1
2011
... ANN has been widely used in many fields of research [113]; this may have been due to ANN attempts to model the functioning of human brains [114]. For example, at present, ANN has become a popular tool for Al-Si alloy performance prediction [[115], [116], [117]]. ANN has been developed to predict the porosity percentage of Al-Si casting alloys and has been used to correlate chemical composition and cooling rate with porosity [118,119]. An ANN has accurately predicted the corrosion resistance of Al-Si-Mg-based metal matrix composites reinforced with SiC particles, with the average square of the Pearson product-moment correlation coefficient (R2), the maximum mean square error, and the minimum root mean squared deviation calculated as 0.9904, 0.00002476, and 0.00157480, respectively. It can be seen that the experimental results are highly consistent with the ANN results [120]. ANN was used to predict the mechanical properties of A356, including yield stress, ultimate tensile strength, maximum force, and elongation percentage; the predictions of the ANN model were found to be in good agreement with experimental data [121,122]. In addition, ANN models have been used to investigate the role of composition and processing parameters in the mechanical properties of microalloyed pipeline steel and to design steel with improved performance with regard to strength, impact toughness, and ductility. Then the models were used as objective functions for multi-objective genetic algorithms to evolve tradeoffs among the conflicting objectives of improved strength, better ductility, and higher impact toughness. Moreover, the Pareto optimal solutions were successfully analyzed to study the role of various parameters in designing pipeline steel with such improved performance [77]. Fig. 4 shows the specific flow chart. ...
1
2012
... ANN has been widely used in many fields of research [113]; this may have been due to ANN attempts to model the functioning of human brains [114]. For example, at present, ANN has become a popular tool for Al-Si alloy performance prediction [[115], [116], [117]]. ANN has been developed to predict the porosity percentage of Al-Si casting alloys and has been used to correlate chemical composition and cooling rate with porosity [118,119]. An ANN has accurately predicted the corrosion resistance of Al-Si-Mg-based metal matrix composites reinforced with SiC particles, with the average square of the Pearson product-moment correlation coefficient (R2), the maximum mean square error, and the minimum root mean squared deviation calculated as 0.9904, 0.00002476, and 0.00157480, respectively. It can be seen that the experimental results are highly consistent with the ANN results [120]. ANN was used to predict the mechanical properties of A356, including yield stress, ultimate tensile strength, maximum force, and elongation percentage; the predictions of the ANN model were found to be in good agreement with experimental data [121,122]. In addition, ANN models have been used to investigate the role of composition and processing parameters in the mechanical properties of microalloyed pipeline steel and to design steel with improved performance with regard to strength, impact toughness, and ductility. Then the models were used as objective functions for multi-objective genetic algorithms to evolve tradeoffs among the conflicting objectives of improved strength, better ductility, and higher impact toughness. Moreover, the Pareto optimal solutions were successfully analyzed to study the role of various parameters in designing pipeline steel with such improved performance [77]. Fig. 4 shows the specific flow chart. ...
1
2016
... Mannodi-Kanakkithodi et al. [123] used a combination of statistical learning and genetic algorithms to predict polymer dielectrics. Within the dataset, the model can not only determine the corresponding properties of any polymer, but can also actively find the specific polymerization that suits its requirements. However, because the model had only seven of the necessary pieces of building-block information, the information guide in the model had certain limitations. ...
1
2014
... Other applications exist for macroscopic performance prediction. Seko et al. [124] used four regression algorithms to predict the melting temperatures of single and binary compounds. Jha et al. [75] predicted polymer glass transition temperatures. Mauro et al. [125] predicted elastic moduli and compressive stress of glass. ...
2
2016
... Other applications exist for macroscopic performance prediction. Seko et al. [124] used four regression algorithms to predict the melting temperatures of single and binary compounds. Jha et al. [75] predicted polymer glass transition temperatures. Mauro et al. [125] predicted elastic moduli and compressive stress of glass. ...
... An adaptive design strategy, tightly coupled with experiments, can accelerate the discovery process by sequentially identifying the next experiments or calculations to effectively navigate the sophisticated search space. The strategy uses inference and global optimization to perform the tradeoff between exploitation and exploration of the search space. Oliynyk et al. [138] used real experimental data, selected a suitable machine learning model, and predicted the model through experiments. Finally, many kinds of intermetallic compounds were discovered. The most crucial finding was RhCd, the first new binary AB compound to be found with a CsCl-type structure in over 15 years. Xue et al. [139] demonstrated this by finding very low thermal hysteresis (DT) NiTi-based shape memory alloys, with Ti50.0Ni46.7Cu0.8Fe2.3Pd0.2 possessing the smallest DT (1.84░K). Wolverton et al. [101] used a general machine learning framework to predict the properties of various materials and discovered a metallic glass alloy. Mauro et al. [125] developed new damage-resistant glasses. Ren et al. [140] found metallic glasses through iterations of machine learning. Yuan et al. [141] used a combination of machine learning and optimization methods to accelerate the discovery of new Pb-free BaTiO3-based piezoelectrics with large electro strains. ...
1
2018
... Pilania et al. [126] used statistical learning methods to quickly and accurately predict the various properties of binary wurtzite superlattices. Toyoura et al. [127] used a machine-learning method called the Gaussian process to efficiently identify low-energy regions that characterize proton conduction in the host lattice. ...
1
2016
... Pilania et al. [126] used statistical learning methods to quickly and accurately predict the various properties of binary wurtzite superlattices. Toyoura et al. [127] used a machine-learning method called the Gaussian process to efficiently identify low-energy regions that characterize proton conduction in the host lattice. ...
1
2017
... To explore the potential thermal runaway problem of lithium-ion electrode particle microstructure, Petrich et al. [128] used a classification model to detect cracks in lithium-ion batteries and applied the method to real electrode data to test its effectiveness. Deringer et al. [129] developed a machine learning-based Gaussian approximation potential model for atomic simulation of liquid and amorphous elemental carbon. Botu et al. [130] proposed a machine learning method called AGNI - an adaptive, generalizable, and neighborhood informed methodology, which can perform fast and quantum accurate mechanical atomic-force prediction in the neighborhood environment of atoms. ...
1
2017
... To explore the potential thermal runaway problem of lithium-ion electrode particle microstructure, Petrich et al. [128] used a classification model to detect cracks in lithium-ion batteries and applied the method to real electrode data to test its effectiveness. Deringer et al. [129] developed a machine learning-based Gaussian approximation potential model for atomic simulation of liquid and amorphous elemental carbon. Botu et al. [130] proposed a machine learning method called AGNI - an adaptive, generalizable, and neighborhood informed methodology, which can perform fast and quantum accurate mechanical atomic-force prediction in the neighborhood environment of atoms. ...
1
2017
... To explore the potential thermal runaway problem of lithium-ion electrode particle microstructure, Petrich et al. [128] used a classification model to detect cracks in lithium-ion batteries and applied the method to real electrode data to test its effectiveness. Deringer et al. [129] developed a machine learning-based Gaussian approximation potential model for atomic simulation of liquid and amorphous elemental carbon. Botu et al. [130] proposed a machine learning method called AGNI - an adaptive, generalizable, and neighborhood informed methodology, which can perform fast and quantum accurate mechanical atomic-force prediction in the neighborhood environment of atoms. ...
1
2012
... Lilienfeld et al. [131] introduced a machine learning model based on nuclear charges and atomic positions to quickly and accurately predict the atomization energy of various organic molecules and proved its applicability for prediction of molecular atomization potential energy curves. Hansen et al. [132] used machine learning to predict molecular atomization energies, which can significantly accelerate the calculation of quantum chemical properties while maintaining high prediction accuracy. Rupp [25] used kernel ridge regression to predict the atomization energies of small organic molecules. Hansen et al. [133] used machine learning (the so-called Bag of Bonds model; BoB) to estimate molecular atomization energies and predict the exact electronic properties of molecules, Fig. 5 shows that BoB is a stronger machine learning model with proper regularization and improved accuracy by simply expanding the molecular database. Moreover, Pilania et al. [33] and Isayev et al. [134] predicted the bandgap energy. ...
1
2013
... Lilienfeld et al. [131] introduced a machine learning model based on nuclear charges and atomic positions to quickly and accurately predict the atomization energy of various organic molecules and proved its applicability for prediction of molecular atomization potential energy curves. Hansen et al. [132] used machine learning to predict molecular atomization energies, which can significantly accelerate the calculation of quantum chemical properties while maintaining high prediction accuracy. Rupp [25] used kernel ridge regression to predict the atomization energies of small organic molecules. Hansen et al. [133] used machine learning (the so-called Bag of Bonds model; BoB) to estimate molecular atomization energies and predict the exact electronic properties of molecules, Fig. 5 shows that BoB is a stronger machine learning model with proper regularization and improved accuracy by simply expanding the molecular database. Moreover, Pilania et al. [33] and Isayev et al. [134] predicted the bandgap energy. ...
2
2015
... Lilienfeld et al. [131] introduced a machine learning model based on nuclear charges and atomic positions to quickly and accurately predict the atomization energy of various organic molecules and proved its applicability for prediction of molecular atomization potential energy curves. Hansen et al. [132] used machine learning to predict molecular atomization energies, which can significantly accelerate the calculation of quantum chemical properties while maintaining high prediction accuracy. Rupp [25] used kernel ridge regression to predict the atomization energies of small organic molecules. Hansen et al. [133] used machine learning (the so-called Bag of Bonds model; BoB) to estimate molecular atomization energies and predict the exact electronic properties of molecules, Fig. 5 shows that BoB is a stronger machine learning model with proper regularization and improved accuracy by simply expanding the molecular database. Moreover, Pilania et al. [33] and Isayev et al. [134] predicted the bandgap energy. ...
... leads to nonrobust fits [133]. 4.3. Fingerprint (descriptor) prediction
If people can effectively learn from available knowledge and data, the material discovery process can be significantly accelerated and simplified. Using a one-dimensional chain system family, Pilania et al. [91] proposed a general form that can discover decision rules and establish a mapping between attributes that are easily accessible to the system and their properties. The results show that fingerprints based on either chemical structure (compositional and configurational information) or electronic charge density distribution can be used for ultra-fast, but accurate property predictions. Harnessing such learning paradigms extends recent efforts to explore systematically huge chemical spaces and can significantly accelerate discovery of new application-specific materials. Curtarolo et al. [135] introduced new material fingerprint descriptors that produce a material mapping network: nodes represent compounds, and connections represent similarities. The mapping network can identify compounds with different physical and chemical properties. Recently, the t-student stochastic neighbor embedding algorithm was used for the first time to extract feature spaces from band-structure ab initio calculations. This algorithm can design different spaces and map them to lower dimensions, enabling simultaneous analysis and exploration of previously unknown band structures for thousands of materials. The more information is available in the material database, the more space can be explored [136]. Ramprasad et al. [137] have studied a series of motif-based topological fingerprints that can represent the major classes of crystals and molecules by numbers. By using a learning algorithm, these fingerprints can be mapped to various properties of crystals and molecules, thereby accelerating property prediction capabilities. Their paper proves the process by simultaneously optimizing two properties, the gap between the highest occupied and lowest unoccupied molecular orbitals EHL and the isotropic polarizability α. The results are shown in Fig. 6 and Table 1, which show that molecules with the required values of EHL and α were obtained. ...
1
2017
... Lilienfeld et al. [131] introduced a machine learning model based on nuclear charges and atomic positions to quickly and accurately predict the atomization energy of various organic molecules and proved its applicability for prediction of molecular atomization potential energy curves. Hansen et al. [132] used machine learning to predict molecular atomization energies, which can significantly accelerate the calculation of quantum chemical properties while maintaining high prediction accuracy. Rupp [25] used kernel ridge regression to predict the atomization energies of small organic molecules. Hansen et al. [133] used machine learning (the so-called Bag of Bonds model; BoB) to estimate molecular atomization energies and predict the exact electronic properties of molecules, Fig. 5 shows that BoB is a stronger machine learning model with proper regularization and improved accuracy by simply expanding the molecular database. Moreover, Pilania et al. [33] and Isayev et al. [134] predicted the bandgap energy. ...
1
2014
... If people can effectively learn from available knowledge and data, the material discovery process can be significantly accelerated and simplified. Using a one-dimensional chain system family, Pilania et al. [91] proposed a general form that can discover decision rules and establish a mapping between attributes that are easily accessible to the system and their properties. The results show that fingerprints based on either chemical structure (compositional and configurational information) or electronic charge density distribution can be used for ultra-fast, but accurate property predictions. Harnessing such learning paradigms extends recent efforts to explore systematically huge chemical spaces and can significantly accelerate discovery of new application-specific materials. Curtarolo et al. [135] introduced new material fingerprint descriptors that produce a material mapping network: nodes represent compounds, and connections represent similarities. The mapping network can identify compounds with different physical and chemical properties. Recently, the t-student stochastic neighbor embedding algorithm was used for the first time to extract feature spaces from band-structure ab initio calculations. This algorithm can design different spaces and map them to lower dimensions, enabling simultaneous analysis and exploration of previously unknown band structures for thousands of materials. The more information is available in the material database, the more space can be explored [136]. Ramprasad et al. [137] have studied a series of motif-based topological fingerprints that can represent the major classes of crystals and molecules by numbers. By using a learning algorithm, these fingerprints can be mapped to various properties of crystals and molecules, thereby accelerating property prediction capabilities. Their paper proves the process by simultaneously optimizing two properties, the gap between the highest occupied and lowest unoccupied molecular orbitals EHL and the isotropic polarizability α. The results are shown in Fig. 6 and Table 1, which show that molecules with the required values of EHL and α were obtained. ...
1
2019
... If people can effectively learn from available knowledge and data, the material discovery process can be significantly accelerated and simplified. Using a one-dimensional chain system family, Pilania et al. [91] proposed a general form that can discover decision rules and establish a mapping between attributes that are easily accessible to the system and their properties. The results show that fingerprints based on either chemical structure (compositional and configurational information) or electronic charge density distribution can be used for ultra-fast, but accurate property predictions. Harnessing such learning paradigms extends recent efforts to explore systematically huge chemical spaces and can significantly accelerate discovery of new application-specific materials. Curtarolo et al. [135] introduced new material fingerprint descriptors that produce a material mapping network: nodes represent compounds, and connections represent similarities. The mapping network can identify compounds with different physical and chemical properties. Recently, the t-student stochastic neighbor embedding algorithm was used for the first time to extract feature spaces from band-structure ab initio calculations. This algorithm can design different spaces and map them to lower dimensions, enabling simultaneous analysis and exploration of previously unknown band structures for thousands of materials. The more information is available in the material database, the more space can be explored [136]. Ramprasad et al. [137] have studied a series of motif-based topological fingerprints that can represent the major classes of crystals and molecules by numbers. By using a learning algorithm, these fingerprints can be mapped to various properties of crystals and molecules, thereby accelerating property prediction capabilities. Their paper proves the process by simultaneously optimizing two properties, the gap between the highest occupied and lowest unoccupied molecular orbitals EHL and the isotropic polarizability α. The results are shown in Fig. 6 and Table 1, which show that molecules with the required values of EHL and α were obtained. ...
3
2015
... If people can effectively learn from available knowledge and data, the material discovery process can be significantly accelerated and simplified. Using a one-dimensional chain system family, Pilania et al. [91] proposed a general form that can discover decision rules and establish a mapping between attributes that are easily accessible to the system and their properties. The results show that fingerprints based on either chemical structure (compositional and configurational information) or electronic charge density distribution can be used for ultra-fast, but accurate property predictions. Harnessing such learning paradigms extends recent efforts to explore systematically huge chemical spaces and can significantly accelerate discovery of new application-specific materials. Curtarolo et al. [135] introduced new material fingerprint descriptors that produce a material mapping network: nodes represent compounds, and connections represent similarities. The mapping network can identify compounds with different physical and chemical properties. Recently, the t-student stochastic neighbor embedding algorithm was used for the first time to extract feature spaces from band-structure ab initio calculations. This algorithm can design different spaces and map them to lower dimensions, enabling simultaneous analysis and exploration of previously unknown band structures for thousands of materials. The more information is available in the material database, the more space can be explored [136]. Ramprasad et al. [137] have studied a series of motif-based topological fingerprints that can represent the major classes of crystals and molecules by numbers. By using a learning algorithm, these fingerprints can be mapped to various properties of crystals and molecules, thereby accelerating property prediction capabilities. Their paper proves the process by simultaneously optimizing two properties, the gap between the highest occupied and lowest unoccupied molecular orbitals EHL and the isotropic polarizability α. The results are shown in Fig. 6 and Table 1, which show that molecules with the required values of EHL and α were obtained. ...
... addressed in the text [137].
Predicted and calculated values of α (in Å3/atom) and EHL (in eV) of the molecules designed from three predicted fingerprints, C, D and E. Data from this table are also shown in the inset of Fig. 6 [137]. ...
... Predicted and calculated values of α (in Å3/atom) and EHL (in eV) of the molecules designed from three predicted fingerprints, C, D and E. Data from this table are also shown in the inset of Fig. 6 [137]. ...
1
2018
... An adaptive design strategy, tightly coupled with experiments, can accelerate the discovery process by sequentially identifying the next experiments or calculations to effectively navigate the sophisticated search space. The strategy uses inference and global optimization to perform the tradeoff between exploitation and exploration of the search space. Oliynyk et al. [138] used real experimental data, selected a suitable machine learning model, and predicted the model through experiments. Finally, many kinds of intermetallic compounds were discovered. The most crucial finding was RhCd, the first new binary AB compound to be found with a CsCl-type structure in over 15 years. Xue et al. [139] demonstrated this by finding very low thermal hysteresis (DT) NiTi-based shape memory alloys, with Ti50.0Ni46.7Cu0.8Fe2.3Pd0.2 possessing the smallest DT (1.84░K). Wolverton et al. [101] used a general machine learning framework to predict the properties of various materials and discovered a metallic glass alloy. Mauro et al. [125] developed new damage-resistant glasses. Ren et al. [140] found metallic glasses through iterations of machine learning. Yuan et al. [141] used a combination of machine learning and optimization methods to accelerate the discovery of new Pb-free BaTiO3-based piezoelectrics with large electro strains. ...
1
2016
... An adaptive design strategy, tightly coupled with experiments, can accelerate the discovery process by sequentially identifying the next experiments or calculations to effectively navigate the sophisticated search space. The strategy uses inference and global optimization to perform the tradeoff between exploitation and exploration of the search space. Oliynyk et al. [138] used real experimental data, selected a suitable machine learning model, and predicted the model through experiments. Finally, many kinds of intermetallic compounds were discovered. The most crucial finding was RhCd, the first new binary AB compound to be found with a CsCl-type structure in over 15 years. Xue et al. [139] demonstrated this by finding very low thermal hysteresis (DT) NiTi-based shape memory alloys, with Ti50.0Ni46.7Cu0.8Fe2.3Pd0.2 possessing the smallest DT (1.84░K). Wolverton et al. [101] used a general machine learning framework to predict the properties of various materials and discovered a metallic glass alloy. Mauro et al. [125] developed new damage-resistant glasses. Ren et al. [140] found metallic glasses through iterations of machine learning. Yuan et al. [141] used a combination of machine learning and optimization methods to accelerate the discovery of new Pb-free BaTiO3-based piezoelectrics with large electro strains. ...
1
2018
... An adaptive design strategy, tightly coupled with experiments, can accelerate the discovery process by sequentially identifying the next experiments or calculations to effectively navigate the sophisticated search space. The strategy uses inference and global optimization to perform the tradeoff between exploitation and exploration of the search space. Oliynyk et al. [138] used real experimental data, selected a suitable machine learning model, and predicted the model through experiments. Finally, many kinds of intermetallic compounds were discovered. The most crucial finding was RhCd, the first new binary AB compound to be found with a CsCl-type structure in over 15 years. Xue et al. [139] demonstrated this by finding very low thermal hysteresis (DT) NiTi-based shape memory alloys, with Ti50.0Ni46.7Cu0.8Fe2.3Pd0.2 possessing the smallest DT (1.84░K). Wolverton et al. [101] used a general machine learning framework to predict the properties of various materials and discovered a metallic glass alloy. Mauro et al. [125] developed new damage-resistant glasses. Ren et al. [140] found metallic glasses through iterations of machine learning. Yuan et al. [141] used a combination of machine learning and optimization methods to accelerate the discovery of new Pb-free BaTiO3-based piezoelectrics with large electro strains. ...
1
2018
... An adaptive design strategy, tightly coupled with experiments, can accelerate the discovery process by sequentially identifying the next experiments or calculations to effectively navigate the sophisticated search space. The strategy uses inference and global optimization to perform the tradeoff between exploitation and exploration of the search space. Oliynyk et al. [138] used real experimental data, selected a suitable machine learning model, and predicted the model through experiments. Finally, many kinds of intermetallic compounds were discovered. The most crucial finding was RhCd, the first new binary AB compound to be found with a CsCl-type structure in over 15 years. Xue et al. [139] demonstrated this by finding very low thermal hysteresis (DT) NiTi-based shape memory alloys, with Ti50.0Ni46.7Cu0.8Fe2.3Pd0.2 possessing the smallest DT (1.84░K). Wolverton et al. [101] used a general machine learning framework to predict the properties of various materials and discovered a metallic glass alloy. Mauro et al. [125] developed new damage-resistant glasses. Ren et al. [140] found metallic glasses through iterations of machine learning. Yuan et al. [141] used a combination of machine learning and optimization methods to accelerate the discovery of new Pb-free BaTiO3-based piezoelectrics with large electro strains. ...
1
2015
... To accelerate the design of new materials, recent studies have screened out many types of promising candidate materials [51], including perovskite compounds [83,142], metal-organic frameworks [143,144], catalysts [145,146], light-emitting molecules [147], and thermoelectric materials [[148], [149], [150], [151]]. A large-scale database based on quantum mechanical calculation of known materials, using machine learning to calculate material screening, is a combination screening process for new materials in unconstrained component space, which can be used for an extensive range of critical material discoveries [87]. In addition, interface structure and energy can be also predicted through virtual screening [152]. ...
1
2012
... To accelerate the design of new materials, recent studies have screened out many types of promising candidate materials [51], including perovskite compounds [83,142], metal-organic frameworks [143,144], catalysts [145,146], light-emitting molecules [147], and thermoelectric materials [[148], [149], [150], [151]]. A large-scale database based on quantum mechanical calculation of known materials, using machine learning to calculate material screening, is a combination screening process for new materials in unconstrained component space, which can be used for an extensive range of critical material discoveries [87]. In addition, interface structure and energy can be also predicted through virtual screening [152]. ...
1
2012
... To accelerate the design of new materials, recent studies have screened out many types of promising candidate materials [51], including perovskite compounds [83,142], metal-organic frameworks [143,144], catalysts [145,146], light-emitting molecules [147], and thermoelectric materials [[148], [149], [150], [151]]. A large-scale database based on quantum mechanical calculation of known materials, using machine learning to calculate material screening, is a combination screening process for new materials in unconstrained component space, which can be used for an extensive range of critical material discoveries [87]. In addition, interface structure and energy can be also predicted through virtual screening [152]. ...
1
2006
... To accelerate the design of new materials, recent studies have screened out many types of promising candidate materials [51], including perovskite compounds [83,142], metal-organic frameworks [143,144], catalysts [145,146], light-emitting molecules [147], and thermoelectric materials [[148], [149], [150], [151]]. A large-scale database based on quantum mechanical calculation of known materials, using machine learning to calculate material screening, is a combination screening process for new materials in unconstrained component space, which can be used for an extensive range of critical material discoveries [87]. In addition, interface structure and energy can be also predicted through virtual screening [152]. ...
1
2016
... To accelerate the design of new materials, recent studies have screened out many types of promising candidate materials [51], including perovskite compounds [83,142], metal-organic frameworks [143,144], catalysts [145,146], light-emitting molecules [147], and thermoelectric materials [[148], [149], [150], [151]]. A large-scale database based on quantum mechanical calculation of known materials, using machine learning to calculate material screening, is a combination screening process for new materials in unconstrained component space, which can be used for an extensive range of critical material discoveries [87]. In addition, interface structure and energy can be also predicted through virtual screening [152]. ...
1
2016
... To accelerate the design of new materials, recent studies have screened out many types of promising candidate materials [51], including perovskite compounds [83,142], metal-organic frameworks [143,144], catalysts [145,146], light-emitting molecules [147], and thermoelectric materials [[148], [149], [150], [151]]. A large-scale database based on quantum mechanical calculation of known materials, using machine learning to calculate material screening, is a combination screening process for new materials in unconstrained component space, which can be used for an extensive range of critical material discoveries [87]. In addition, interface structure and energy can be also predicted through virtual screening [152]. ...
1
2016
... To accelerate the design of new materials, recent studies have screened out many types of promising candidate materials [51], including perovskite compounds [83,142], metal-organic frameworks [143,144], catalysts [145,146], light-emitting molecules [147], and thermoelectric materials [[148], [149], [150], [151]]. A large-scale database based on quantum mechanical calculation of known materials, using machine learning to calculate material screening, is a combination screening process for new materials in unconstrained component space, which can be used for an extensive range of critical material discoveries [87]. In addition, interface structure and energy can be also predicted through virtual screening [152]. ...
1
2016
... To accelerate the design of new materials, recent studies have screened out many types of promising candidate materials [51], including perovskite compounds [83,142], metal-organic frameworks [143,144], catalysts [145,146], light-emitting molecules [147], and thermoelectric materials [[148], [149], [150], [151]]. A large-scale database based on quantum mechanical calculation of known materials, using machine learning to calculate material screening, is a combination screening process for new materials in unconstrained component space, which can be used for an extensive range of critical material discoveries [87]. In addition, interface structure and energy can be also predicted through virtual screening [152]. ...
1
2015
... To accelerate the design of new materials, recent studies have screened out many types of promising candidate materials [51], including perovskite compounds [83,142], metal-organic frameworks [143,144], catalysts [145,146], light-emitting molecules [147], and thermoelectric materials [[148], [149], [150], [151]]. A large-scale database based on quantum mechanical calculation of known materials, using machine learning to calculate material screening, is a combination screening process for new materials in unconstrained component space, which can be used for an extensive range of critical material discoveries [87]. In addition, interface structure and energy can be also predicted through virtual screening [152]. ...
1
2016
... To accelerate the design of new materials, recent studies have screened out many types of promising candidate materials [51], including perovskite compounds [83,142], metal-organic frameworks [143,144], catalysts [145,146], light-emitting molecules [147], and thermoelectric materials [[148], [149], [150], [151]]. A large-scale database based on quantum mechanical calculation of known materials, using machine learning to calculate material screening, is a combination screening process for new materials in unconstrained component space, which can be used for an extensive range of critical material discoveries [87]. In addition, interface structure and energy can be also predicted through virtual screening [152]. ...
1
2016
... To accelerate the design of new materials, recent studies have screened out many types of promising candidate materials [51], including perovskite compounds [83,142], metal-organic frameworks [143,144], catalysts [145,146], light-emitting molecules [147], and thermoelectric materials [[148], [149], [150], [151]]. A large-scale database based on quantum mechanical calculation of known materials, using machine learning to calculate material screening, is a combination screening process for new materials in unconstrained component space, which can be used for an extensive range of critical material discoveries [87]. In addition, interface structure and energy can be also predicted through virtual screening [152]. ...
1
2018
... At present, the scale of data that can be applied to machine learning in the field of materials is still very small, making it impossible to generate rigorous predictions of material properties and new material discoveries through machine learning. The predicted results are often only close to the probability values of real data and cannot be truly used for experimental guidance. Even if some experts propose to improve machine learning application methods for small-scale datasets, for example by adding a rough attribute estimation to the feature space to build a machine learning model using a small-scale material dataset, the accuracy of the prediction can be improved [153]. However, even with the gradual development of machine learning and deep learning algorithms, from the application experience of each algorithm model, the scale of the dataset is still the main factor in model prediction accuracy. This problem is reflected not only in the machine learning algorithms, but also in deep learning, which has attracted much attention in recent years. In the last few decades, it has been found that material data are presented relatively intact in a large number of scientific papers, and literature data extraction has made some progress in many specific fields, such as chemistry [154] and biomedicine [[155], [156], [157]]. Some studies using these material data have already been carried out. For example, a paper information-assisted extraction software package has been developed and is available online (https://www.mgedata.cn/) [158]. Xie et al. [159] designed the composition of high-performance copper alloys using machine learning based on data from the literature. Edward et al. [42] obtained material synthesis information from academic publications. Kononova et al. [160] obtained inorganic material synthesis recipes by using text mining of scientific publications. However, data in the literature are rarely used by material scientists, and even if the data are used, the data scale is small because there is no mature method to extract data from the material literature. So far, no relatively complete corpus has been developed in the materials field to support the application of material data. Therefore, in the materials genome initiative, material prediction through machine learning should not only focus on research into the machine learning algorithm itself, but should also extract valuable material data from the materials science literature. This would involve first classifying and pre-processing these literature data, then using machine learning algorithms to predict the performance data of interest, and finally finding the correlations among material composition, process, structure, and performance. These steps will play a key role in discovering new properties of materials. ...
1
2017
... At present, the scale of data that can be applied to machine learning in the field of materials is still very small, making it impossible to generate rigorous predictions of material properties and new material discoveries through machine learning. The predicted results are often only close to the probability values of real data and cannot be truly used for experimental guidance. Even if some experts propose to improve machine learning application methods for small-scale datasets, for example by adding a rough attribute estimation to the feature space to build a machine learning model using a small-scale material dataset, the accuracy of the prediction can be improved [153]. However, even with the gradual development of machine learning and deep learning algorithms, from the application experience of each algorithm model, the scale of the dataset is still the main factor in model prediction accuracy. This problem is reflected not only in the machine learning algorithms, but also in deep learning, which has attracted much attention in recent years. In the last few decades, it has been found that material data are presented relatively intact in a large number of scientific papers, and literature data extraction has made some progress in many specific fields, such as chemistry [154] and biomedicine [[155], [156], [157]]. Some studies using these material data have already been carried out. For example, a paper information-assisted extraction software package has been developed and is available online (https://www.mgedata.cn/) [158]. Xie et al. [159] designed the composition of high-performance copper alloys using machine learning based on data from the literature. Edward et al. [42] obtained material synthesis information from academic publications. Kononova et al. [160] obtained inorganic material synthesis recipes by using text mining of scientific publications. However, data in the literature are rarely used by material scientists, and even if the data are used, the data scale is small because there is no mature method to extract data from the material literature. So far, no relatively complete corpus has been developed in the materials field to support the application of material data. Therefore, in the materials genome initiative, material prediction through machine learning should not only focus on research into the machine learning algorithm itself, but should also extract valuable material data from the materials science literature. This would involve first classifying and pre-processing these literature data, then using machine learning algorithms to predict the performance data of interest, and finally finding the correlations among material composition, process, structure, and performance. These steps will play a key role in discovering new properties of materials. ...
1
2012
... At present, the scale of data that can be applied to machine learning in the field of materials is still very small, making it impossible to generate rigorous predictions of material properties and new material discoveries through machine learning. The predicted results are often only close to the probability values of real data and cannot be truly used for experimental guidance. Even if some experts propose to improve machine learning application methods for small-scale datasets, for example by adding a rough attribute estimation to the feature space to build a machine learning model using a small-scale material dataset, the accuracy of the prediction can be improved [153]. However, even with the gradual development of machine learning and deep learning algorithms, from the application experience of each algorithm model, the scale of the dataset is still the main factor in model prediction accuracy. This problem is reflected not only in the machine learning algorithms, but also in deep learning, which has attracted much attention in recent years. In the last few decades, it has been found that material data are presented relatively intact in a large number of scientific papers, and literature data extraction has made some progress in many specific fields, such as chemistry [154] and biomedicine [[155], [156], [157]]. Some studies using these material data have already been carried out. For example, a paper information-assisted extraction software package has been developed and is available online (https://www.mgedata.cn/) [158]. Xie et al. [159] designed the composition of high-performance copper alloys using machine learning based on data from the literature. Edward et al. [42] obtained material synthesis information from academic publications. Kononova et al. [160] obtained inorganic material synthesis recipes by using text mining of scientific publications. However, data in the literature are rarely used by material scientists, and even if the data are used, the data scale is small because there is no mature method to extract data from the material literature. So far, no relatively complete corpus has been developed in the materials field to support the application of material data. Therefore, in the materials genome initiative, material prediction through machine learning should not only focus on research into the machine learning algorithm itself, but should also extract valuable material data from the materials science literature. This would involve first classifying and pre-processing these literature data, then using machine learning algorithms to predict the performance data of interest, and finally finding the correlations among material composition, process, structure, and performance. These steps will play a key role in discovering new properties of materials. ...
1
2013
... At present, the scale of data that can be applied to machine learning in the field of materials is still very small, making it impossible to generate rigorous predictions of material properties and new material discoveries through machine learning. The predicted results are often only close to the probability values of real data and cannot be truly used for experimental guidance. Even if some experts propose to improve machine learning application methods for small-scale datasets, for example by adding a rough attribute estimation to the feature space to build a machine learning model using a small-scale material dataset, the accuracy of the prediction can be improved [153]. However, even with the gradual development of machine learning and deep learning algorithms, from the application experience of each algorithm model, the scale of the dataset is still the main factor in model prediction accuracy. This problem is reflected not only in the machine learning algorithms, but also in deep learning, which has attracted much attention in recent years. In the last few decades, it has been found that material data are presented relatively intact in a large number of scientific papers, and literature data extraction has made some progress in many specific fields, such as chemistry [154] and biomedicine [[155], [156], [157]]. Some studies using these material data have already been carried out. For example, a paper information-assisted extraction software package has been developed and is available online (https://www.mgedata.cn/) [158]. Xie et al. [159] designed the composition of high-performance copper alloys using machine learning based on data from the literature. Edward et al. [42] obtained material synthesis information from academic publications. Kononova et al. [160] obtained inorganic material synthesis recipes by using text mining of scientific publications. However, data in the literature are rarely used by material scientists, and even if the data are used, the data scale is small because there is no mature method to extract data from the material literature. So far, no relatively complete corpus has been developed in the materials field to support the application of material data. Therefore, in the materials genome initiative, material prediction through machine learning should not only focus on research into the machine learning algorithm itself, but should also extract valuable material data from the materials science literature. This would involve first classifying and pre-processing these literature data, then using machine learning algorithms to predict the performance data of interest, and finally finding the correlations among material composition, process, structure, and performance. These steps will play a key role in discovering new properties of materials. ...
1
2013
... At present, the scale of data that can be applied to machine learning in the field of materials is still very small, making it impossible to generate rigorous predictions of material properties and new material discoveries through machine learning. The predicted results are often only close to the probability values of real data and cannot be truly used for experimental guidance. Even if some experts propose to improve machine learning application methods for small-scale datasets, for example by adding a rough attribute estimation to the feature space to build a machine learning model using a small-scale material dataset, the accuracy of the prediction can be improved [153]. However, even with the gradual development of machine learning and deep learning algorithms, from the application experience of each algorithm model, the scale of the dataset is still the main factor in model prediction accuracy. This problem is reflected not only in the machine learning algorithms, but also in deep learning, which has attracted much attention in recent years. In the last few decades, it has been found that material data are presented relatively intact in a large number of scientific papers, and literature data extraction has made some progress in many specific fields, such as chemistry [154] and biomedicine [[155], [156], [157]]. Some studies using these material data have already been carried out. For example, a paper information-assisted extraction software package has been developed and is available online (https://www.mgedata.cn/) [158]. Xie et al. [159] designed the composition of high-performance copper alloys using machine learning based on data from the literature. Edward et al. [42] obtained material synthesis information from academic publications. Kononova et al. [160] obtained inorganic material synthesis recipes by using text mining of scientific publications. However, data in the literature are rarely used by material scientists, and even if the data are used, the data scale is small because there is no mature method to extract data from the material literature. So far, no relatively complete corpus has been developed in the materials field to support the application of material data. Therefore, in the materials genome initiative, material prediction through machine learning should not only focus on research into the machine learning algorithm itself, but should also extract valuable material data from the materials science literature. This would involve first classifying and pre-processing these literature data, then using machine learning algorithms to predict the performance data of interest, and finally finding the correlations among material composition, process, structure, and performance. These steps will play a key role in discovering new properties of materials. ...
1
2019
... At present, the scale of data that can be applied to machine learning in the field of materials is still very small, making it impossible to generate rigorous predictions of material properties and new material discoveries through machine learning. The predicted results are often only close to the probability values of real data and cannot be truly used for experimental guidance. Even if some experts propose to improve machine learning application methods for small-scale datasets, for example by adding a rough attribute estimation to the feature space to build a machine learning model using a small-scale material dataset, the accuracy of the prediction can be improved [153]. However, even with the gradual development of machine learning and deep learning algorithms, from the application experience of each algorithm model, the scale of the dataset is still the main factor in model prediction accuracy. This problem is reflected not only in the machine learning algorithms, but also in deep learning, which has attracted much attention in recent years. In the last few decades, it has been found that material data are presented relatively intact in a large number of scientific papers, and literature data extraction has made some progress in many specific fields, such as chemistry [154] and biomedicine [[155], [156], [157]]. Some studies using these material data have already been carried out. For example, a paper information-assisted extraction software package has been developed and is available online (https://www.mgedata.cn/) [158]. Xie et al. [159] designed the composition of high-performance copper alloys using machine learning based on data from the literature. Edward et al. [42] obtained material synthesis information from academic publications. Kononova et al. [160] obtained inorganic material synthesis recipes by using text mining of scientific publications. However, data in the literature are rarely used by material scientists, and even if the data are used, the data scale is small because there is no mature method to extract data from the material literature. So far, no relatively complete corpus has been developed in the materials field to support the application of material data. Therefore, in the materials genome initiative, material prediction through machine learning should not only focus on research into the machine learning algorithm itself, but should also extract valuable material data from the materials science literature. This would involve first classifying and pre-processing these literature data, then using machine learning algorithms to predict the performance data of interest, and finally finding the correlations among material composition, process, structure, and performance. These steps will play a key role in discovering new properties of materials. ...
1
2019
... At present, the scale of data that can be applied to machine learning in the field of materials is still very small, making it impossible to generate rigorous predictions of material properties and new material discoveries through machine learning. The predicted results are often only close to the probability values of real data and cannot be truly used for experimental guidance. Even if some experts propose to improve machine learning application methods for small-scale datasets, for example by adding a rough attribute estimation to the feature space to build a machine learning model using a small-scale material dataset, the accuracy of the prediction can be improved [153]. However, even with the gradual development of machine learning and deep learning algorithms, from the application experience of each algorithm model, the scale of the dataset is still the main factor in model prediction accuracy. This problem is reflected not only in the machine learning algorithms, but also in deep learning, which has attracted much attention in recent years. In the last few decades, it has been found that material data are presented relatively intact in a large number of scientific papers, and literature data extraction has made some progress in many specific fields, such as chemistry [154] and biomedicine [[155], [156], [157]]. Some studies using these material data have already been carried out. For example, a paper information-assisted extraction software package has been developed and is available online (https://www.mgedata.cn/) [158]. Xie et al. [159] designed the composition of high-performance copper alloys using machine learning based on data from the literature. Edward et al. [42] obtained material synthesis information from academic publications. Kononova et al. [160] obtained inorganic material synthesis recipes by using text mining of scientific publications. However, data in the literature are rarely used by material scientists, and even if the data are used, the data scale is small because there is no mature method to extract data from the material literature. So far, no relatively complete corpus has been developed in the materials field to support the application of material data. Therefore, in the materials genome initiative, material prediction through machine learning should not only focus on research into the machine learning algorithm itself, but should also extract valuable material data from the materials science literature. This would involve first classifying and pre-processing these literature data, then using machine learning algorithms to predict the performance data of interest, and finally finding the correlations among material composition, process, structure, and performance. These steps will play a key role in discovering new properties of materials. ...
1
2019
... At present, the scale of data that can be applied to machine learning in the field of materials is still very small, making it impossible to generate rigorous predictions of material properties and new material discoveries through machine learning. The predicted results are often only close to the probability values of real data and cannot be truly used for experimental guidance. Even if some experts propose to improve machine learning application methods for small-scale datasets, for example by adding a rough attribute estimation to the feature space to build a machine learning model using a small-scale material dataset, the accuracy of the prediction can be improved [153]. However, even with the gradual development of machine learning and deep learning algorithms, from the application experience of each algorithm model, the scale of the dataset is still the main factor in model prediction accuracy. This problem is reflected not only in the machine learning algorithms, but also in deep learning, which has attracted much attention in recent years. In the last few decades, it has been found that material data are presented relatively intact in a large number of scientific papers, and literature data extraction has made some progress in many specific fields, such as chemistry [154] and biomedicine [[155], [156], [157]]. Some studies using these material data have already been carried out. For example, a paper information-assisted extraction software package has been developed and is available online (https://www.mgedata.cn/) [158]. Xie et al. [159] designed the composition of high-performance copper alloys using machine learning based on data from the literature. Edward et al. [42] obtained material synthesis information from academic publications. Kononova et al. [160] obtained inorganic material synthesis recipes by using text mining of scientific publications. However, data in the literature are rarely used by material scientists, and even if the data are used, the data scale is small because there is no mature method to extract data from the material literature. So far, no relatively complete corpus has been developed in the materials field to support the application of material data. Therefore, in the materials genome initiative, material prediction through machine learning should not only focus on research into the machine learning algorithm itself, but should also extract valuable material data from the materials science literature. This would involve first classifying and pre-processing these literature data, then using machine learning algorithms to predict the performance data of interest, and finally finding the correlations among material composition, process, structure, and performance. These steps will play a key role in discovering new properties of materials. ...






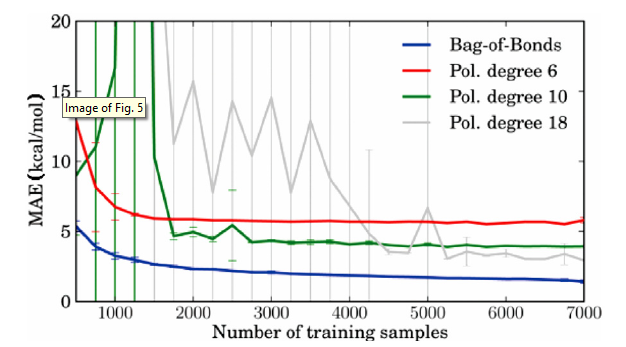
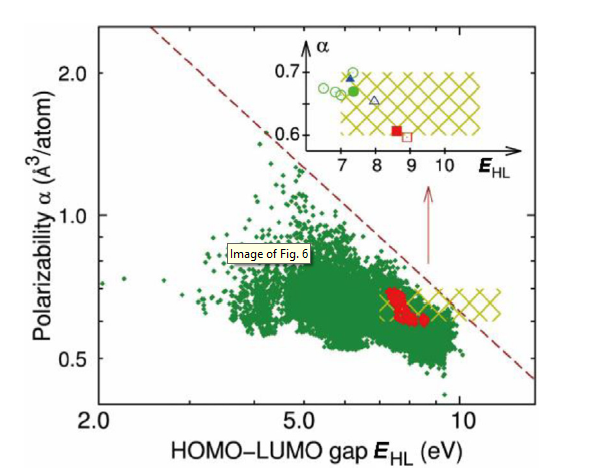



 WeChat
WeChat
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}