

1. Introduction
Excessive consumptions of fossil sources such as oil, coal and natural gas led to the over-emission of carbon dioxide, causing serious of consequences: global warming, climate change and energy shortage, which broke the global ecological balance. Conversion of CO2 into valuable chemicals (methanol, formic acid, ethanol and olefins, etc.) [[1], [2], [3], [4], [5]], not only effectively alleviates the climate change and greenhouse effect contributed from carbon emissions, but also provides new opportunities for the utilization of carbon-based chemicals and sustainable carbon economic [6]. Methanol, as an alternative addition of gasoline and diesel fuels and feedstocks for synthetic of olefins and ethers, has been widely used in chemical industry, pharmaceutical and energy field [7]. At present, Cu-ZnO based catalysts are the most efficient scheme to produce methanol from syngas [8,9]. However, due to thermodynamic and kinetic limitations, the hydrogenation process is always carried out at high temperature and pressure. And there are still drawbacks such as low methanol selectivity and rapid deactivations during the syngas hydrogenation to methanol [10].
Based on the deficiency of energy consumption and activity performance of CO2 direct hydrogenation, Milstein and coworkers proposed a new approach of indirect conversion of CO2 to methanol via hydrogenation of carbonates, and carbonates derivatives can be produced from CO2 fixation under mild conditions [[11], [12], [13]]. Using Ru and Mn homogeneous catalysts, it is easy to realize high activity and selectivity of methanol under relative low pressure and temperature, which achieved the comprehensive utilization of CO2 to alcohol [14,15]. Although it is an efficient and atomic economy alternative route (Scheme 1), the disadvantage of product separation over the homogeneous catalyst prevents the scale-up application. From the viewpoints of green chemistry and durability of catalyst, a serious of heterogeneous catalysts were developed for the valid hydrogenation of carbonates, such as Cu/HMS [16], CuCr2O4 [17] and Cu/CeO2 [18], etc. Glucose modified Cu8G1/SiO2-AE catalyst prepared by ammonia evaporation method showed enhanced catalytic performance, furnishing selectivity to methanol and EG at almost full conversion of EC, with a turnover frequency (TOF) value for methanol formation about 39 h-1 [19]. A hierarchically core/shell-structured Silicalite-1@Cu composite was also developed via a base-assisted chemo-selective host-guest interaction between the silicon species of MFI-type Silicalite-1 and external Cu salt source. The S-1@Cu hybrid possessed the co-existence of Cu°/Cu+ active species, which served as a highly active sites for the selective EC hydrogenation [20]. However, these catalysts have drawbacks such as low selectivity of alcohol and deactivation at highly LHSV during the reaction. Thus, construction of stable catalyst with high LHSV is essential for the improvement of methanol and diols production.
Scheme 1.
Scheme 1.
Methanol production for the CO2 indirect utilization via carbonate process.
Owing to the unique catalytic feature, copper-based catalysts have been widely applied for the vapor-phase selective hydrogenation of carbon-oxygen bond, such as CO, dimethyl oxalate and carbonates [[21], [22], [23]]. As a potential alternative for the conventional hydrogenation catalysts, Cu/SiO2 catalyst exhibited satisfying catalytic activity [24]. However, quick deactivation and little resistance against sintering have prevented the scale-up application of Cu/SiO2 catalysts [25]. Thus, considerable efforts focused on the electronic and structure modification of Cu/SiO2 catalysts have been done to enhance the durability of copper-based catalytic under high LHSV [26]. Cobalt has been employed as a promoter for Cu/HMS catalyst in the hydrogenation of dimethyl oxalate (DMO). The catalytic activity could be significantly enhanced because of the facilitated hydrogen dissociation and optimized copper species dispersion. Au, Ag also attracted much attention as promoters for the regulation of bimetals synergetic effect of the hydrogenation performance [27]. The EG yield of a certain amount of Ag incorporated Cu/SiO2 catalyst exhibited 2.2 times higher than that of monometallic catalyst [28]. However, modification of catalysts for the hydrogenation of carbonates has been rarely investigated. Therefore, architecture of metal promoted copper-based catalysts will extend the application of Cu/SiO2 catalysts in hydrogenation procedure.
Herein, we have developed an Ag-modified copper phyllosilicate (CuPs) catalyst which maintained superior activity in the hydrogenation of EC, and provided high methanol and EG yield at variety ranges of EC LHSV. With the co-ammonia evaporation hydrothermal method, a small amount of Ag decorated catalyst showed unique physicochemical features. Even at 2.0 h-1 LHSV of EC, the obtained catalyst remained about totally conversion of EC, and high yield of methanol as well as EG. Characterizations demonstrated that silver-affected electronic and structural regulation was essential for the catalytic performance.
2. Experimental
2.1. Catalyst preparation
The ultra-thin copper phyllosilicate nanotubes were prepared by an ammonia evaporation hydrothermal method based on the previous report [21]. Briefly, 1.0 g of P123 was added in 100 mL of deionized and vigorously stirred for 2 h to sufficiently dissolve at room temperature. 11.292 g of Cu(NO3)2·3H2O was added over 1.0 wt.% P123 solution with continuous stirring. Then aqueous ammonia was slowly added dropwise, adjusting the pH value to 10∼11, and dark blue solution was obtained. Then, 10 g 30 wt% silica-gel was mixed with the obtained copper ammonia solution and kept continuously stirring at room temperature overnight to get a suspension system. The beaker was placed into a water bath, warmed to 363 K and kept 2∼3 h. A light blue colloid was acquired and transferred into autoclave Teflon reactor to proceed the hydrothermal treatment at 473 K for 36 h. The obtained precipitation was washed with water and ethanol alternately to give a pale blue solid and dried overnight at 373 K, then calcined at 723 K for 4 h in static air at heating rate of 10 K/min. The obtained sample was marked as CuPs catalyst.
CuPs Ag-copre catalyst was prepared by a procedure similar with the fabrication of ultra-thin copper phyllosilicate nanotubes. The specific step was that 0.346 g of AgNO3 was simultaneously dissolved with copper precursor and hydrothermal treated for the next step. The as-obtained sample was referred as CuPs Ag-copre catalyst.
CuPs Ag-post catalyst was fabricated by post modification of CuPs nanotubes. Briefly, the as-prepared CuPs nanotubes were re-dispersed in 100 mL of deionized water. 0.346 g of AgNO3 was added and ultrasonic immersed for 30 min. Then impregnation was ended with water vapored out. Other dried and calcined steps were the same as the above procedure, and the as-obtained catalyst was marked as CuPs Ag-post.
Ag/SiO2 catalyst was also prepared via an ammonia-evaporation (AE) method. 0.350 g of AgNO3 dissolved in 100 mL of deionized water. Then concentrated ammonia aqueous solution was slowly added dropwise to adjust the pH value to 10∼11. 10 g of silica sol was added to the silver-ammonia solution, and continuously stirred at room temperature for 12 h. The beaker was placed in a water bath and kept at 363 K for several hours until the pH was neutral. The as-obtained precipitation was washed with water and ethanol alternately to give precipitation and further dried overnight in an oven at 373 K, and calcined at 723 K in static air. The sample was referred as Ag/SiO2.
Cu/SiO2 catalyst was fabricated by wet impregnating method with Cu(NO3)2·3H2O on silica. The drying and calcine conditions were the same as the procedure of CuPs catalyst, which was denoted as Cu/SiO2.
2.2. Catalyst characterization
Specific surface areas of the samples were measured using N2 physisorption at 77 K on a Micromeritics TriStar 3020 apparatus, the specific surface areas were calculated following the Brunauer-Emmett-Teller (BET) method. Pore size distributions were calculated by the BJH method.
The wide-angle XRD scanning studies were conducted on a Bruker D8 Advance X-ray diffractometer using nickel-filtered Cu Kα radiation with 2θ range of 10-90°, a scanning speed of 4°/min, a voltage and current of 40 kV and 40 mA, respectively. The full width at half maximum (FWHM) of Cu (111) reflection was acquired for calculating crystallite sizes by using the Scherrer equation.
Fourier transform infrared spectroscopy (FT-IR) experiments of all the catalysts were performed by using a Bruker Vector 22 spectrometer equipped with a DTGS detector and a KBr beam splitter.
TEM and SEM micrographs were obtained by transmission electron microscopy (TEM; JEOL JEM 2010) and scanning electron microscope (SEM, Nova NanoSem 450).
The surface species were investigated by X-ray photoelectron spectroscopy (XPS; RBD 147 upgraded Perkin Elmer PHI 5300). The spectrum was recorded with Mg Kα line as the excitation source (1253.6 eV). All binding energies were calibrated with contaminant carbon (284.6 eV) with an uncertainty of ±0.1 eV. XPSPeak4.1 software was employed to deconvolute the peaks using the Shirley-type baseline.
The reducibility of the calcined samples was evaluated by H2 temperature-programmed reduction (H2-TPR) on a Tianjin XQ TP5080 auto-adsorption apparatus. 20 mg of the catalyst was outgassed at 473 K under Ar flow for 1.5 h. After cooling to room temperature, the in-line gas was switched to 5 vol.% H2/Ar, with a ramping rate of 10 K/min to a final temperature of 773 K. Hydrogen consumption was simultaneously monitored by a thermal conductivity detector (TCD). The dispersion and metallic copper surface areas of the catalysts were determined by N2O chemisorption at 333 K using a procedure described in the literature [29].
Firstly, a flow of 5 vol.% H2/Ar (30 mL/min) was used for the reduction of copper species, and the temperature was raised from room temperature to 573 K over 2 h with a heating rate of 10 K/min. Secondly, the procedure was initiated after the reduced sample was cooled to 333 K in Ar (30 mL/min) and purged until the baseline being smooth. Then, pure N2O (20 mL/min) was introduced to the catalyst at 333 K for 0.5 h. Subsequently, the sample was purged with Ar (20 mL/min) for 0.5 h to remove the residual N2O. Thirdly, a flow of 5 vol.% H2/Ar (30 mL/min) was also used for the reduction of Cu2O surface species, and the temperature was raised from room temperature to 773 K with a heating rate of 10 K/min. By assuming spherical shape of copper metal particles and 1.46 × 1019 atomsCu/m2, the dispersion of copper (DCu) and copper surface area (SCu) can be calculated as follows:
Hydrogen consumption: A1
Hydrogen consumption: A2
$D_{Cu}(\%)=\frac{2{{A}_{2}}}{{{A}_{1}}}\times 100$
$S_{Cu}(m^{2}·g_{cat-1})=\frac{D_{Cu}\times N_{av}\times W_{Cu}}{M_{Cu}\times 1.46\times 10^{19}}$
Nav: Avogadro constant,
WCu (wt. %): the content of copper in the catalyst,
MCu: relative atomic weight.
2.3. Catalytic activity evaluation
The catalytic performance of as-prepared catalysts was evaluated using a fixed-bed reactor equipped with a stainless-steel tubular (length × outer diameter × inner diameter = 500 × 20 × 10 mm). Typically, 1.0 g of the sample (40-60 mesh) was packed in the center of reactor tube with quartz powder loaded to fill both sides. Under the condition of good tightness, the catalyst was reduced under flow H2 (0.5 MPa, 50 mL/min) at 573 K for 4 h, with a ramping rate of 2 K/min. After the catalyst bed was cooled to reaction temperature, EC solution was fed into the tubular reactor using a high-pressure current pump under H2 flow (150 mL/min) and system pressure of 3.0 MPa. The condensed products were collected after 6 h pre-reaction and analyzed by gas chromatograph (GC-2010 plus, Shimadzu Co., Ltd.) with RT-Wax capillary column (60 m × 0.32 mm ×0.50 μm) and a flame-ionization detector (FID).
3. Results and discussion
3.1. Catalytic performance
The catalytic performance for hydrogenation of EC to methanol and EG at 453 K and 3.0 MPa H2 was evaluated over the as-prepared catalysts. In the reaction, one equivalent of methanol and EG were produced from hydrogenation of one mole of carbonyl and dioxygen-ethyl in ethylene carbonate with gas by-products of CO and CO2 liberated [30], wherein the presented activity basis on carbonyl corresponded to chemical fixed CO2 was shown in Table 1. CuPs catalyst had a preferable nanotube which could confine the copper particles, thus exhibited high conversion (>99%) and high yield of methanol (72%) and EG (98%), respectively. Ag modified CuPs catalyst provided higher yield of products than CuPs. Under relatively mild conditions, CuPs Ag-copre could acquire >99% EC conversion and 79% methanol yield, and CuPs Ag-post could still obtain 74% methanol yield, as well as 99% EG yield. Therefore, modification of Ag could be regarded as a preferable choice for the enhancement over copper-based catalyst in the hydrogenation reactions. Ag/SiO2 catalyst synthesized from AE method was also investigated for the production of methanol, and the catalytic performance turned out to be not well (Table 1, Entry 9), which revealed that different decoration methods would promote the production of methanol. For the CuPs Ag-copre catalyst with the best modification, the total yield of methanol could reach up to 79%. The as-prepared CuPs Ag-copre catalyst revealed certainly catalytic performance improvement compared with previous works in the selective hydrogenation of carbonates (Table S1). Additionally, Cu/SiO2 obtained from impregnation method showed a lower EC conversion (47%) and methanol yield (28%), indicating that the excellent catalytic performance probably relied on the specific structure of CuPs catalyst.
Table 1 Catalytic activity over Ag modified CuPs catalysts.
| Entry | Catalyst | Conversion (%) | Yield (%) | |
|---|---|---|---|---|
| Methanol | Ethylene glycol | |||
| 1 | CuPs | >99 | 72 | 98 |
| 2 | CuPs Ag-copre | >99 | 79 | 99 |
| 3 | CuPs Ag-post | >99 | 74 | 98 |
| 4 | CuPs Fe-copre | 96 | 31 | 87 |
| 5 | CuPs Ni-copre | >99 | 12 | 85 |
| 6 | CuPs Co-copre | 92 | 42 | 79 |
| 7 | CuPs Ru-copre | >99 | 73 | 99 |
| 8 | Cu/SiO2a | 47 | 28 | 36 |
| 9 | Ag/SiO2 | 11 | 6 | 9 |
Reaction Conditions: LHSV(EC) =0.20 h-1, P(H2) =3.0 MPa, T =453 K, n(H2):n(EC) = 80.
a Cu/SiO2 catalyst prepared by impregnation method.
Under similar reaction conditions, different metal promoters were applied for the catalytic hydrogenation reaction (Table 1), such as Fe, Ni, Co and Ru. Among these metal additions, CuPs Ni-copre catalyst performed the minimum methanol yield and high selectivity of gas products. As an ideal active metal for gas reforming, Ni has a strong C—C dissociation ability [31]. In the hydrogenation of EC, C—C and C—O bonds of ethylene carbonate were cracked, thus large amounts of gas phase by-products were formed. Ru decoration was conducive to the formation of methanol, due to the improved hydrogen dissociation ability of ruthenium, which demonstrated that the activation ability of hydrogen was responsible for the improvement of methanol production.
The catalytic activity at different EC LHSV was investigated over catalysts with different Ag loading orders. As shown in Fig. 1, under 453 K and 3.0 MPa H2 conditions, when LHSV was varied from 0.10 to 2.00 h-1, all the catalysts could approximately maintain 99% EC conversion. Whereas, methanol yield over CuPs Ag-copre catalyst decreased to 75% with LHSV increasing to 1.50 h-1. Both CuPs and CuPs Ag-post catalysts showed similar decreasing tendency in the range of 1.50-2.00 h-1 EC, and the rapid deactivation of CuPs catalyst is obvious. As an exothermic reaction, contact time between substrates and active sites was a key factor for the hydrogenation reaction under low LHSV condition. Lots of latent heats were taken out along with the feed gas and products. But the residence time of EC was so short during the reaction at high LHSV. The substrates could not sufficiently contact with the catalysts, thus reduced the conversion of EC. After the decoration of Ag, the catalyst had higher LHSV and obtained high methanol yield compared with CuPs catalyst, indicating that the metal incorporation could efficiently increase the operation window range of LHSV. The long-term running operation was evaluated under the conditions of 453 K and 3.0 MPa. No obvious decrease of EC conversion and methanol yield was reserved even after 150 h time on stream (Fig. 2), which indicated the CuPs Ag-copre catalyst acquired stabilized catalytic performance for the hydrogenation of EC.
Fig. 1.
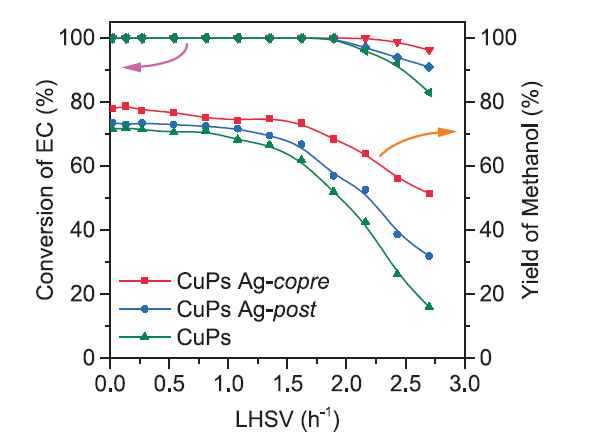
Fig. 1.
EC conversion and methanol yield as a function of different EC LHSV over Ag modified CuPs catalysts.
Fig. 2.
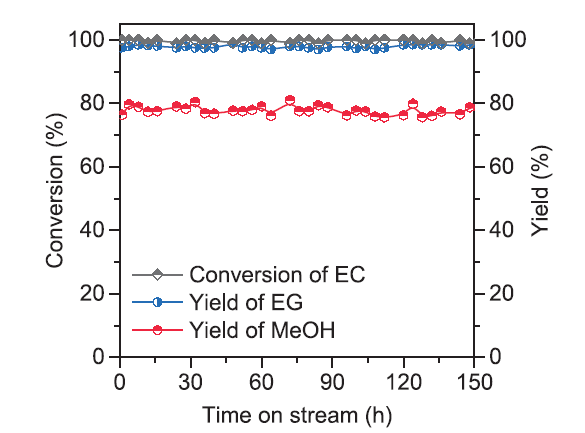
Fig. 2.
EC hydrogenation over CuPs Ag-copre catalyst as a function of time on stream. Reaction Conditions: LHSV(EC) =0.20 h-1, P(H2) =3.0 MPa, T =453 K, n(H2):n(EC) = 80.
3.2. Catalyst characterizations
3.2.1. Textural properties of the catalysts
After the hydrothermal treatment at 473 K for 36 h, the as-prepared catalysts exhibited different N2 adsorption-desorption isothermals. As shown in Fig. 3, the CuPs catalyst showed a high specific surface area of 497 m2/g. The isothermal of CuPs Ag-copre catalyst did not change so much, compared with calcined monometallic CuPs sample, but the specific surface area gradually increased to 511 m2/g. The surface area of calcined CuPs Ag-post sample synthesized by post modification decreased to 408 m2/g, indicating that the surface properties of the catalysts were optimized by the addition of silver. [32,33] Pore volume of the catalysts changed along with the specific surface area. Wherein the co-ammonia evaporation hydrothermal method derived catalyst resulted in the increase of pore volume up to 1.21 cm3/g, followed by CuPs and CuPs Ag-post, which showed pore volume of 1.05 and 0.53 cm3/g, respectively. Hence, the decoration of Ag in CuPs Ag-copre catalyst did not change the physical properties so much. However, Ag specie would fill part of the columnar pores of the CuPs precursor, which led to the decrease of pore volume. In addition, all the catalysts showed Langmuir IV type of isothermal as shown in Fig. 3(a). And there was quite large difference in the type of hysteresis loop. CuPs Ag-copre and CuPs Ag-post acted as H3 type and H2b type hysteresis loops, respectively. CuPs, the most probable diameter of CuPs Ag-copre and CuPs Ag-post turned out to be 3.2, 3.1 and 12.0 nm respectively. A new pore size distribution at 12.0 nm appeared after modification of Ag. The structure of the catalyst Ag/SiO2 was also further illustrated. The specific surface area, pore volume, and average pore size were 137 m2/g, 0.45 cm3/g and 10.3 nm, which demonstrated that Ag/SiO2 contained disordered stacked pore.
Fig. 3.

Fig. 3.
N2 adsorption-desorption isotherms (a) and pore diameter distribution (b) of the calcined CuPs Ag-copre, CuPs Ag-post, CuPs and Ag/SiO2 samples.
3.2.2. FT-IR
Fig. 4 showed Fourier-transform IR (FT-IR) spectra of the calcined catalysts. The surface hydroxyl groups vibration (δOH) located at 670 cm-1 were observed, as well as the shoulder peak (νSiO) at frequency of 1040 cm-1, which were assigned to the structure of copper phyllosilicate species. The stretching bands appearing at 800 and 1110 cm-1 could be ascribed to the symmetric and asymmetric vibration of νSiO, suggesting the presence of portion Si-O-Si. [34] Thus, certain amount of copper phyllosilicate and SiO2 coexisted in the as-calcined samples. And it should be noticed that the integrated δOH band of CuPs Ag-copre decreased compared with CuPs catalyst, meaning that the addition of Ag regulated the silicate layer structure or reduced the relative content of copper phyllosilicate species. However, the Ag impregnated CuPs Ag-post catalyst showed similar vibration features, especially the almost vanished symmetric band of νSiO, which could be ascribed to the destroy of the lamellar structure by ultrasonic treatment.
Fig. 4.
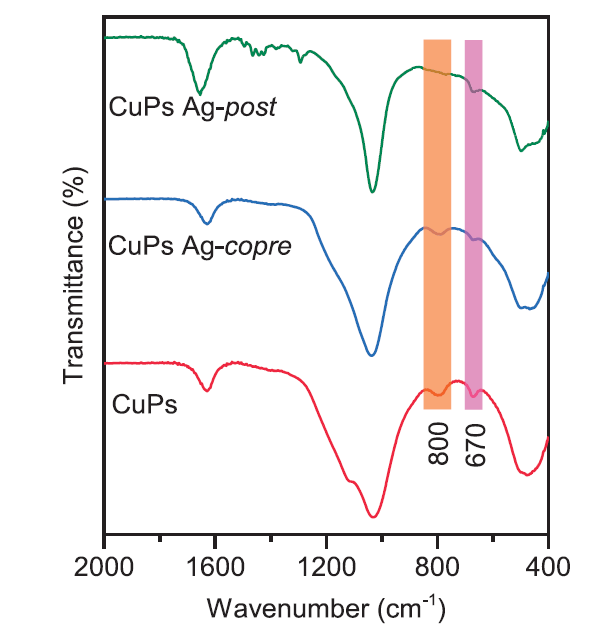
Fig. 4.
FT-IR spectra of the calcined Ag modified CuPs catalysts.
3.2.3. XRD, TEM and SEM
The XRD patterns of calcined and reduced samples were shown in Fig. 5. The calcined CuPs and CuPs Ag-copre catalyst exhibited analogous diffractions. The features 2θ located at 19.9, 21.8, 30.8, 35.0, 57.5 and 62.4°, which could attribute the catalyst to chrysocolla (JCPDS No.: 27-0188, Cu2-xSi2O5(OH)3·xH2O). [35] Meanwhile, no other unclassified diffractions appeared, which indicated that the copper phyllosilicate was of high purity. For the calcined CuPs Ag-post sample, diffractions of 2θ at 35.5, 38.7 and 48.6° were detected and corresponded to tenorite (JCPDS No.: 48-1548, CuO), revealing that the silver ions partially exchanged with the octahedral [CuO] layer in silicates through ultrasonic and thermal impregnation [36]. The dissociative copper ions and extra silver ions redeposited on the surface of copper phyllosilicate nanotubes, which resulted in CuO crystalline particles.
Fig. 5.

Fig. 5.
XRD patterns of the Ag modified CuPs catalysts: (a) calcined, (b) reduced.
In the reduced catalysts, strong diffractions at 2θ of 43.3, 50.4 and 74.1° demonstrated the formation of face-centered cubic (fcc) copper (JCPDS No.: 04-0836). In particular, stretched diffractions of Cu2O (JCPDS No.: 05-0667) at 36.4 and 61.3° assigned to the (111) (220) lattice were inspected. The peak at 22° came from amorphous SiO2. All the as-prepared catalysts contained similar phase compositions, but the CuPs Ag-copre catalysts have gained higher content of Cu2O than other samples. According to the Scherrer formula, the crystalline sizes of copper species over CuPs, CuPs Ag-copre and CuPs Ag-post catalysts were 6.2, 7.5 and 9.3 nm (Table 2), respectively. The increased copper grain size of CuPs Ag-post catalyst further proved that deposited CuO particles were not conducive to the size control of copper species. It was worth noting that the reduced CuPs Ag-post catalyst showed a diffraction at 38.1°, which was corresponded to Ag (111) plane (JCPDS No.: 04-0783), revealing the formation of Ag particles and also resulted in the phase separation of Ag and Cu. It was interesting to find that diffractions of Ag species were undetected for the CuPs Ag-copre catalyst with similar content of Ag loading.
Table 2 Physicochemical parameters of the Ag modified CuPs catalysts.
| Catalyst | a M loading (%) | SBET (m2/g) | Vp (cm3/g) | dp (nm) | bdCu (nm) | cDCu (%) | cSCu0 (m2/gcat) | d TOF (h-1) | |
|---|---|---|---|---|---|---|---|---|---|
| Cu | Ag | ||||||||
| CuPs | 33.5 | - | 497 | 1.05 | 6.6 | 6.2 | 27.8 | 60.5 | 9.1 |
| CuPs Ag-copre | 35.1 | 1.6 | 511 | 1.21 | 7.0 | 7.5 | 33.1 | 75.6 | 12.7 |
| CuPs Ag-post | 31.9 | 2.5 | 408 | 0.53 | 4.2 | 9.3 | 26.1 | 54.3 | 11.3 |
| Ag/SiO2 | - | 1.9 | 137 | 0.45 | 10.3 | 7.7 | - | - | - |
a Metal loading determined by ICP-OES.
b Cu average diameter of particle size calculated from Cu(111) FWHM of XRD results using Scherrer equation.
c Cu dispersion and surface area of Cu0 calculated from H2-TPR and N2O titration.
d TOF is defined as the methanol generating per Cu site per time.
The morphologies and structures of CuPs Ag-copre and CuPs Ag-post catalysts were observed using transmission electron microscopy (TEM) and scanning electron microscopy (SEM). Long-lengthen nanotubes with diameter of 4.3 nm, and 0.8 nm wall thickness were observed in the calcined CuPs Ag-copre catalyst (Fig. 6(a)), along with an open mouth at the wire top. When compared with the lattice spacing of the copper phyllosilicate, the ultra-thin wall was indexed to few layers of silicates. Nanoparticles could be uniformly distributed on the surface of the nanotubes after the post introduction of Ag. This feature further suggested the ion exchange and precipitation procedure of extra copper and silver species [37].
Fig. 6.

Fig. 6.
TEM images of CuPs Ag-copre (a,c) and CuPs Ag-post (b,d) catalysts: calcined (a,b) and reduced (c,d); SEM images of the calcined CuPs Ag-copre (e) and CuPs Ag-post (f) samples.
In the reduced catalysts, copper nanoparticles were dispersed or embedded in the nanotubes, and all the samples exhibited an approximate average size distribution. And the specific structure of copper phyllosilicate offered a confined space, efficiently restricting the growth of copper particles. With different order of Ag decoration, and the as-prepared catalysts exhibited mean copper particle size of 6.8 and 7.5 nm. SEM was employed for the morphology view, CuPs Ag-copre was most likely the structure of interwoven wires (Fig. 6). Post impregnation treatment led to the length diminution of nanotubes and even partial block of the tube cavity. The results further demonstrated the different preparation methods had certainly influence on the nanotube structure.
In addition, the XRD and TEM characterizations were further conducted for checking the structure changes of spent CuPs Ag-copre catalyst after being used for 150 h. As shown in Fig. S1, it could be observed that diffractions of Cu species slightly enhanced compared with the fresh reduced CuPs Ag-copre catalyst, revealing the crystalline size of Cu enlarged under reduction conditions. TEM image further confirmed the growth up of copper particles, and the average size was 8.1 nm. These results demonstrated the specific cavity and confined space of CuPs nanotube retarded the active species agglomeration efficiently. And the catalyst showed good stability in the hydrogenation process under reaction conditions.
3.2.4. TPR
H2-TPR was conducted to investigate the effects of modification methods for catalyst redox property and metal support interaction. Asymmetric hydrogen consumption appeared in CuPs catalyst, corresponding to the sequential reduction of Cu2+ to Cu+ and Cu+ to Cu° for copper phyllosilicate from the deconvolution results (Fig. 7). Whereas, CuPs Ag-copre H2 consumption shifted to low temperature region, meantime peak area assigned to Cu+ to Cu° species at 542 K increased, which indicated that the decoration of Ag enhanced the reducibility of copper species. Silver played a positive role in promoting the reduction peak symmetry, meaning that copper species were well-dispersed with narrow distributions. CuPs Ag-post sample showed a shoulder consumption at 564 K. Combined with the TEM images, the peak could be attributed to the precipitated or redeposited copper and silver species from ion-exchange process. The N2O titration method was further performed to calculate the dispersion of copper species and Cu° surface area after the incorporation of silver. The dispersion of copper species over CuPs Ag-copre and CuPs Ag-post samples were 33.1 and 26.1%, respectively, and the corresponding Cu surface areas (SCu) were 75.6 and 54.3 m2/g. Such results further proved that co-ammonia evaporation hydrothermal process had effectively optimized the dispersion of copper species and interaction between metal and the support. In addition, the H2-TPR profile of the referenced Ag/SiO2 catalyst also exhibited a weak hydrogen consumption peak at 510 K. Huang et. al have reported that in Ag modified Cu/SiO2 catalyst synthesized by using urea-assisted gelation method, the reduction peak gradually shifted to lower temperature with the introduction of Ag species. The coherent of Cu and Ag attributed to the improvement of catalysts reducibility [28]. The present result agreed well with the observed trend of copper dispersion and Cu° surface area.
Fig. 7.
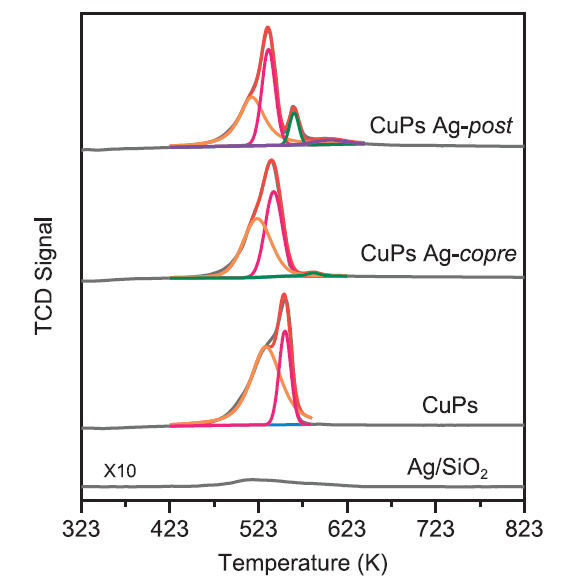
Fig. 7.
H2-TPR profiles of the Ag modified CuPs catalysts.
3.2.5. XPS and XAES
The chemical state and valence distribution of the catalyst surface elements were tested by XPS, and results were shown in Fig. 8. It could be confirmed that the Cu 2p3/2 core level binding energy of the as-reduced CuPs Ag-copre, CuPs Ag-post and CuPs catalysts were all around 932.7 eV. Meanwhile, no obvious peaks and satellites presented at 934.9 and 945.0 eV, which should be assigned to Cu2+ for 2p→3d transition, demonstrating that copper species were converted to valence of Cu+ and Cu0 under hydrogen reduction at 573 K. Nevertheless, the binding energy of Cu 2p3/2 slightly migrated to higher regions with addition of Ag. To further investigate the state of Ag species, Ag 3d XPS experiments were carried out and shown in Fig. 8(c). Interestingly, Ag species partially penetrated into the copper phyllosilicate layers, and no obviously Ag 3d signal at the range of 364∼379 eV could be found for CuPs Ag-copre catalyst. On the contrary, metallic Ag 3d5/2 signal appeared at 368.4 eV in CuPs Ag-post catalyst, indicating that Ag species deposited on the surface of catalyst. Binding energy shift also appeared for Ag 3d core level compared with Ag/SiO2 catalyst. Thus, we speculated that charge transfer occurred between the coherent copper and silver species. As reported, Ag species acted as a negative electronic site to stabilize the positive Cu+ under reducing H2 atmosphere.
Fig. 8.

Fig. 8.
Cu 2p (a), Ag 3d (b) XPS and Cu LMM (c) XAES spectra of the reduced Ag modified CuPs catalysts.
Since binding energy of Cu+ and Cu° species cannot be differed by only 0.1∼0.2 eV distinction in Cu 2p orbital, Auger electron spectroscopy (AES) was employed to discriminate copper species with Cu LMM peak deconvolution, and the results were shown in Fig. 8(b). The Auger kinetic energy of Cu+ and Cu° were around 914.6 and 917.8 eV respectively. It also could be observed the distribution of Cu+ and Cu° species was different in the as-reduced catalysts. Wherein the CuPs catalyst without Ag addition contained relative low content of Cu+ species (17.2%), and the CuPs Ag-post catalyst showed moderate content of Cu+ (24.8%). The catalyst synthesized by the co-ammonia evaporation hydrothermal method exhibited the content of Cu+ as high as 30.1% (Table S2). The reason for valence distribution might be attributed to the ion exchange and formation of CuAg alloy, and part of independent Ag species distributed on the copper particles surface (Fig. 6). For the CuPs Ag-copre catalyst, copper and silver coherent species were dispersed uniformly as bimetallic alloy or cluster. Therefore, silver species had certainly affected the properties of adjacent Cu+ due to its negative electronic feature, which could obtain and stabilize a relatively higher Cu+ content in the catalyst.
3.3. Structure-performance relationship
Modified copper phyllosilicate nanotubes prepared with different incorporation orders of Ag exhibited different catalytic performance and physicochemical properties, and CuPs Ag-copre catalyst achieved enhanced activity for the hydrogenation of EC to methanol and EG. The XRD patterns and TEM images showed that ion exchange of Ag with CuPs took place after post addition of silver ions, which resulted in partial surface damage of CuPs nanotubes. Excess copper and silver species were deposited on the surface of CuPs nanotubes. Finally, copper and silver species were detached in the reduced CuPs Ag-post catalyst. Meanwhile, CuPs Ag-copre catalyst showed no obvious phase and structure differences, compared with monometallic CuPs. And catalyst with Ag permeating in silicate layer was obtained. The coherent interaction between copper and silver species enhanced the reducibility. N2O titration results also revealed that the dispersion of copper species on the CuPs Ag-copre catalyst was slightly higher than the other two samples, thus indicating that co-ammonia evaporation hydrothermal treatment led to a stronger metal support interaction. It has been demonstrated that suitable zinc incorporated CuO-ZnO/SiO2 catalyst resulted in higher Cu+ content on the surface of catalysts, improved dispersion of Cu° particles and sintering resistance [38]. Silver doped Cu/HMS catalyst obtained via co-ammonia evaporation method also indicated appropriate silver was used to achieve intermetallic nanoparticles, and the interaction between Ag and Cu increased the ratio of Cu+, which enhanced the adsorption of substrate and promoted the catalytic performance [39]. In a word, Ag species were appropriately embedded into the layers of silicates under ammonia evaporation hydrothermal treatment, which might enhance the hydrogen dissociation ability at lower reduction temperature, and improved the reduction and activation process of copper species. Although the silver species in CuPs Ag-post catalyst has also enhanced the reducibility of copper too, the structural defects caused by the evolution of some copper ions and deposition have reduced the dispersion of copper species, as well large copper metal was not conducive to selective hydrogenation of carbonyl [40]. Whereas, CuPs Ag-copre showed higher dispersion of Cu and Cu° surface area than the unmodified catalyst. It has been proved that increasing of Cu surface area enhanced the hydrogen activation ability of the catalyst, which was beneficial to the hydrogenation of carbonates reactions. Although the Cu° surface area of CuPs Ag-post declined certain amount slightly, the added Ag species alleviated the disadvantages of copper species for hydrogen dissociation. Thus the moderate catalytic activity still could be obtained. As well, it can be observed that the catalytic activity of CuPs Ag-copre was higher than that of the unmodified monometallic catalyst at high value of LHSV. The presence of Ag species also stabilized Cu+ species on the catalyst surface. For a better comparison of the catalytic activities, the turnover frequency (TOF) value was also normalized based on the copper surface area (SCu) and shown in Table 2. The CuPs Ag-copre catalyst presented a higher TOF of 12.7 h-1 compared with CuPs Ag-post (11.4 h-1) and CuPs (9.1 h-1) samples, which was mainly attributed to the higher ratio of Cu+ specie.
Poels studied Zn and Mn decorated Cu/SiO2 catalysts for the hydrogenation performance of ethyl acetate. By controlling Cu0 surface area, Zn or Mn were homogeneously deposited on the catalyst surface, efficiently adjusting the strength of surface metal-oxygen bonds, thereby inhibiting the selectivity of side-product alkanes [41]. It also showed that appropriate amount of Ag [28] and Au [42,43] acted as a promoter for Cu species dispersion and regulation of Cu+ species in Cu/SiO2 and stabilized the ratio of valence distribution of Cu+/Cu0. Meanwhile, the sintering and agglomeration of the copper nanoparticles were retarded, thus the stability of the catalyst was enhanced. When Pt was used as an auxiliary reagent, the H2 activation ability of the catalyst was significantly enhanced. Meanwhile, Cu2+ species reduction process was accelerated. The reduced Cu and Pt species form an alloy structure, which also modulated the ratio of Cu+/Cu0, and thus exhibited high ethylene selectivity. In addition, metal and non-metal oxides such as lanthanum oxide [44] and boric acid [45,46] were used for the dispersion of copper species and the relative content of Cu+ in order to modulate the electronic properties of the catalyst surface and the interaction between copper species and carriers. As reported, Cu0 and Cu+ sites were essential for the H2 decomposition and carbonyl oxygen bond cleavage, and balanced active Cu species had potential contributions in the hydrogenation process. Both species had significant influence on the structure regulation, which accounted for the excellent catalytic performance. As an electron accepter, Ag could optimize the valence distribution of Cu+ and Cu0 on the catalyst surface, resulted in the improvement of the catalytic performance (Fig. 9).
Fig. 9.
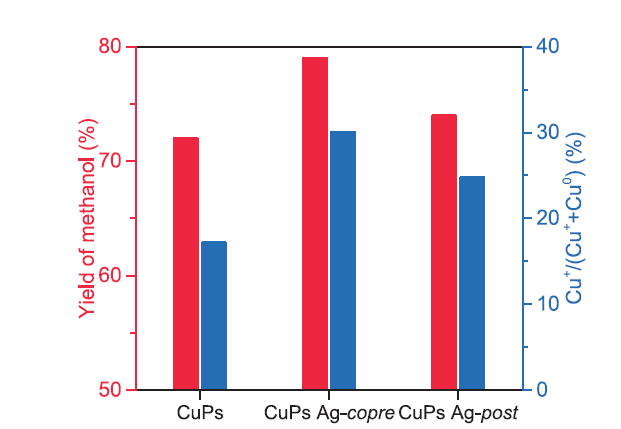
Fig. 9.
Correlation of methanol yield and Cu+ content over Ag modified CuPs catalysts.
4. Conclusion
In summary, the rapid deactivation of Cu/SiO2 catalysts at a high EC LHSV has been an important obstacle for industry application. Herein, silver modified copper phyllosilicates nanotubes were fabricated by different strategies, and the CuPs Ag-copre catalyst synthesized by co-ammonia evaporation hydrothermal process achieved the catalytic performance of 79% methanol yield and 99% glycol yield in a series of reaction ranges of EC LHSV. In the ion-exchange process, a large amount of copper ions and excess silver species were dissolved in the solution, while the copper oxide and silver particles were deposited around the CuPs nanotubes, which resulted in the existence of copper oxide of CuPs Ag-post catalyst. The silver species inlaid copper phyllosilicate layers of CuPs Ag-copre catalyst effectively promoted the reducibility and dispersion of copper species effectively. Meanwhile, the distribution of Cu+ and Cu0 species on the catalyst surface was also regulated. The Ag and Cu coherent species exhibited crucial contributions to the enhancement of metal and support interaction. Furthermore, the electronic state and structural property were optimized for the conversion of carbonate, which increased the stability of the long-term running of the catalyst. In addition, this method could also be applied in the enhancement of copper-based catalysts using other metal or metal oxide promotor.
Acknowledgements
This work was financially supported by the Natural Science Foundation of Shanghai (19ZR1403500), National Natural Science Foundation of China (No. 21373054) and the Natural Science Foundation of Shanghai Science and Technology Committee (No. 08DZ2270500).
Appendix A. Supplementary data
Supplementary material related to this article can be found, inthe online version, at doi: https://doi.org/10.1016/j.jmst.2020.02.020.
Reference
DOI
URL
PMID
[Cited within: 1]

One of the main stumbling blocks in developing rational design strategies for heterogeneous catalysis is that the complexity of the catalysts impairs efforts to characterize their active sites. We show how to identify the crucial atomic structure motif for the industrial Cu/ZnO/Al(2)O(3) methanol synthesis catalyst by using a combination of experimental evidence from bulk, surface-sensitive, and imaging methods collected on real high-performance catalytic systems in combination with density functional theory calculations. The active site consists of Cu steps decorated with Zn atoms, all stabilized by a series of well-defined bulk defects and surface species that need to be present jointly for the system to work.
DOI
URL
PMID
[Cited within: 1]
Catalytic reforming provides a practical technique for on-board hydrogen production in fuel cell applications. The high energy density, easy transportation and non-toxicity of biomass-derived dimethyl ether (bio-DME) offer potential to replace methanol for on-board steam reforming (SR). Presently, the reaction mechanism over conventional Cu-based SR catalysts remains elusive, limiting the rational design of highly efficient reforming systems. Herein, we build a catalytic system for bio-DME SR with dual-sites of Cu species, i.e., Cu(+) and Cu(0) sites, and achieve a record-high H2 production rate of 1145 mol kgcat (-1) h(-1). Via regulating the ratios of the dual-sites of Cu, we clearly describe molecular understandings on SR. And we discover that the substantially boosted activity is induced by a new Cu(+)-determined reaction path substituting the conventional Cu(0)-determined path. Intrinsically, Cu2O can act as a physical spacer and hydroxyl consumer to suppress the aggregation of metallic Cu species in SR. Due to the unique structure of metallic Cu surrounded by Cu2O, the catalyst exhibits robust catalytic performance even after severe thermal treatment. These findings open a new avenue for designing efficient catalytic reforming systems with commercial potential.
DOI
URL
PMID
[Cited within: 1]
The oxidation of the endocrine disruptor, diethylstilbestrol (DES) in aqueous media by ultrasound is significantly enhanced by Fe(II) catalyst. The observed enhancement is likely the result of increased levels of hydroxyl radicals from the iron-promoted reduction of the hydrogen peroxide produced during ultrasonic irradiation. The degradation is effective over a range of concentrations and is consistent with pseudo first-order kinetics. Relatively high concentrations of hydrogen peroxide, ~450 mM, are present in solution under our experimental conditions after 1 h of ultrasonic irradiation (665 kHz). The concentration of H2O2 in solution decreased with the addition of Fe(II) along with an increase in the degradation of DES. Hydrogen peroxide alone does not appreciably degrade DES. Our results demonstrate ultrasonic-induced degradation of DES can be accelerated with the addition of Fe(II). The combination of ultrasonic irradiation and Fe(II)-promoted conversion of H2O2 to hydroxyl radical may provide a valuable strategy for the treatment of organic pollutants.

 WeChat
WeChat
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}