Copyright:
2020 Editorial board of Journal of Materials Science & Technology Copyright reserved, Editorial board of Journal of Materials Science & Technology
More
Abstract
We report first-principles results of the point defect properties in a V-Ta-Cr-W high-entropy alloy (HEA) with the body-centered cubic (bcc) structure. Different from the widely-investigated face-centered cubic (fcc) HEAs, the local lattice distortion is more pronounced in bcc ones, which has a strong influence on the defect properties and defect evolution under irradiation. Due to the large size of Ta, the exchange between vacancies and Ta exhibits lower energy barriers. On the other hand, interstitial dumbbells containing V and Cr possess lower formation energies. These defect energetics predicts an enrichment of V and Cr and a depletion of Ta and W in the vicinity of defect sinks. Besides, we find that interstitial dumbbells favor the [110] orientation in the HEA, instead of [111] direction in most nonmagnetic bcc metals, which helps to slow down interstitial diffusion significantly. Consequently, the distribution of migration energies for vacancies and interstitials exhibit much larger overlap regions in the bcc HEA compared to fcc HEAs, leading to the good irradiation resistance by enhancing defect recombination. Our results suggest that HEAs with the bcc structure may bear excellent irradiation tolerance due to the particular defect properties.
ShijunZhao. Defect properties in a VTaCrW equiatomic high entropy alloy (HEA) with the body centered cubic (bcc) structure[J]. Journal of Materials Science & Technology, 2020, 44(0): 133-139 https://doi.org/10.1016/j.jmst.2019.10.025
1. Introduction
Tungsten (W) is a leading candidate material as the divertor or the plasma-facing materials (PFMs) in future fusion reactors, owing to its high melting temperature, high thermal conductivity, low sputtering erosion rates, and small tritium retention [[1], [2], [3]]. Despite these advantages, W suffers from its poor mechanical properties due to its low fracture toughness and high brittle to ductile transition temperature (BDTT), which severely restricts its operation temperature window. Besides, irradiation from He ions that can be produced by fusion neutrons may induce significant morphological changes in the surface of W, such as the formation of nanometer-scale bubbles and low-density fuzz structures [2]. These adverse effect has greatly impeded the application of W in fusion reactors. Therefore, various pathways have been proposed and investigated in order to improve the performance of W, mainly focusing on adding minor dopant elements or tailoring its microstructures [4,5].
Different from these extensively adopt routes, the development of high-entropy alloy (HEAs) provides another powerful way to tune alloy properties through engineering the chemical disorder in alloys. HEAs are multicomponent alloys composed of different elemental species all at high concentrations [[6], [7], [8]]. Due to the high configurational entropy of mixing, these alloys tend to crystallize as solid solutions in a simple underlying face-centered cubic (fcc) or body-centered cubic (bcc) structures. In HEAs, the local atomic environment of every atom is unique, so that defect properties vary even in the same material. In particular, defect energies, including their formation and migration energies, exhibit distributions [9,10]. As a result, the mutual interaction between vacancies and interstitials may get enhanced, which contributes to the improved defect recombination and accordingly, irradiation resistance [11].
Indeed, recent experimental results have demonstrated outstanding radiation resistance in W-based HEAs [12,13]. For example, the W38Ta36Cr15V11 alloy with the bcc structure shows no sign of irradiation-created dislocation loops after ion irradiation up to a dose of 8 displacement per atom (dpa) [13]. The good radiation resistance of the V-Ta-Cr-W alloy is explained by the formation of Cr- and V-rich second-phase precipitate as well as the equal mobility of point defects which favors defect recombination. Besides the promising irradiation performance, it is reported that W-based bcc HEAs exhibit superior mechanical properties at high temperatures compared to Ni-based superalloys [14]. By tuning the fabrication strategy, both yield strength and ductility of these bcc HEAs can be improved [15], making them potential candidate materials for future nuclear applications. More importantly, previous experiments indicate that bcc HEAs almost exhibit no radiation hardening effect [13,16], in contrast to the noticeable radiation hardening observed in previous fcc HEAs. These findings closely relate to the specific distribution of defects generated by energetic ion bombardment. Therefore, it is of paramount significance to evaluate the defect properties in bcc HEAs. Most previous studies about defect properties are focused on HEAs with the fcc structures [9,10,[17], [18], [19]], and their properties in bcc HEAs still remain unexplored.
In this work, we present a first-principles study of the defect properties in a bcc HEA, i.e. VTaCrW. We first investigate the lattice distortion of the HEA, which is found to be more pronounced compared to previous fcc HEAs. The formation and migration energies of vacancies are then calculated, and their distributions are presented. We find smaller migration barriers for Ta vacancies due to the large atomic size of Ta. Because of the large lattice distortion, interstitials in the HEA can only be formed at some particular locations. We have analyzed the structures of the stable interstitial dumbbells and show that most dumbbells are along the [110] directions. These results are helpful for the understanding of the irradiation performance of this novel bcc HEAs.
2. Method
Ab initio calculations were carried out based on the density functional theory (DFT), using pseudopotentials generated within the projector augmented wave (PAW) approach [20] as implemented in the Vienna ab initio simulation package [21,22]. The exchange-correlation interactions were described by the generalized gradient approximation (GGA) of the Perdew-Burke-Ernzerhof (PBE) form [23]. The Methfessel-Paxton smearing [24] with a smearing width of 0.1 eV was employed to account for partial occupancies of the metallic HEA systems. Periodic boundary conditions and the supercell approach was used for all calculations. The cutoff energy of plane waves was 300 eV. For the defect formation and migration energy calculations, a 4 × 4×4 bcc supercell containing 128 atoms was used and the Brillouin zone was sampled with a 3 × 3×3 k-point mesh based on the Monkhorst-Pack scheme. The convergence threshold for the total energy and atomic force were 10-4 eV and 10-2 eV/A, respectively. The chemical disorder of the HEAs was modelled using special quasirandom structures (SQS) that were constructed by optimization of the Warren-Cowley short range order (SRO) parameters [25,26] using a Monte-Carlo simulated annealing algorithm [27]. The volume of the supercell was fixed during ionic relaxation; the volume was fitted from the calculated energy-volume relations.
The defect formation energy is calculated from:
Ef=Ed-E0±μd (1)
where Ed and E0 are the energies of defective and defect-free supercells respectively, and μd is the chemical potential of defect species d. The signs of + and - correspond to a vacancy and an interstitial, respectively. In this work, the elemental chemical potentials were estimated following our previous method [9,10,28].
The migration barrier of defects is calculated by directly optimizing the saddle configurations, as explained in our previous studies [10]. Different from fcc HEAs investigated previously, the bcc equiatomic WTaCrV alloy considered here exhibit rather large lattice distortions (more details given below). Consequently, some types of defects are not stable upon relaxation at some locations, especially for interstitial defects. Due to this fact, the saddle configuration can barely be found for interstitials, and only the migration barriers of vacancies are considered. In this case, the saddle structure contains a lattice atom locating in the middle of two nearest vacancies. By fixing the position of this atom, we calculate the energy of this saddle structure as Es, and the barrier can then be obtained from Em=Es-Ed, where Ed is the energy of the system containing the initial vacancy.
3. Result
3.1. Local lattice distortion
For the VTaCrW alloy, we first calculate its equilibrium lattice constant based on the obtained energy-volume relation. The result is 3.102 Å, which is in the range of the lattice constants of the four corresponding pure constituent metals (3.04 Å for V, 3.31 Å for Ta, 2.85 Å for Cr, and 3.18 Å for W [29]). Based on the calculated result, we calculate the formation enthalpy of this HEA to be 0.11 eV/formula at 0 K. After a full relaxation, it is found that the magnetic moment diminished in this system, even if spin-polarized calculations are performed. Therefore, nonmagnetic calculations were carried out for all the following calculations.
From the equilibrium lattice structure, we have studied the atomic displacement of different elements. Based on a Voronoi tessellation analysis [30], the averaged atomic volume for each element can be deduced. The results are provided in Table 1, which shows that the size of these elements is in the order Cr < V<W < Ta. This order is consistent with their atomic volumes in their pure bcc bulk metals. The difference in the atomic volumes is as large as 1 Å3, which is much larger compared to fcc HEAs.
Table 1
Table 1 Averaged atomic volume calculated through the Voronoi tessellation method based on the SQS supercell.
The distributions of the first and second nearest neighbor (nn) distances are given in Fig. 1(a) and (b). We also calculate the radial displacement of all the atoms as shown in Fig. 1(c), using the perfect bcc lattice as a reference. Consistent with the trend of atomic volumes, the 1nn distance between V and Cr are smaller than those between W and Ta. The distribution of 1nn and 2nn distance shows a slightly overlap region, suggesting the large local lattice distortion effect. The calculated 1nn distances are larger than those reported in fcc HEAs [9]. According to a previous analysis of lattice distortion, the α2 parameter for the studied HEA is 0.04, which is higher than most values of the solid solutions reported in the same paper [31]. This is also an indication of the large lattice distortion in the studied HEA. On the other hand, the radial deviation distance does not exactly follow the trend of atomic size. For example, the larger W exhibits a smaller deviation than the smaller V. The average atomic displacements are 0.094, 0.094, 0.079, and 0.077 Å for V, Ta, Cr, and W respectively. The smaller relaxation displacement of W in the pristine structure is also found in the defected structures, as detailed in the next section.
Fig. 1. Local lattice distortion in the studied HEA: (a) distribution of the first nearest neighbor distance; (b) distribution of the second nearest neighbor distance; (c) radial deviation distance of all the atoms. In (a) and (b), 1 denotes for V, 2 for Ta, 3 for Cr, and 4 for W.
The different displacement of these elements relates to their different electronic structures. In particular, the studied VTaCrW alloy includes both 3d metals (V and Cr) and 5d metals (Ta and W). It has been shown that the bonding between 3d‒5d metals has stronger covalent character than 5d‒5d metals [32], which affects the relaxation properties of elements. The averaged partial density of states (PDOS) of d orbitals for each element is given in Fig. 2. It shows that 3d Cr and V exhibit a large DOS value at the Fermi energy (N(EF)), compared to 5d Ta and W, an indication of the lattice instability of Cr and V. Indeed, relaxation helps to lower N(EF), thus stabilizing the lattice. This factor may contribute to the large displacement of V and Cr. The Fermi energy locates in the pseudogap of the PDOS, separating the t2g and eg states. Relaxation leads to a shift of the low-energy t2g states toward left, causing a larger separation of the t2g and eg states. The PDOS of Ta and W are very similar. Nevertheless, the displacement of Ta is much larger than W, which suggests that other factors also come to play in determining their relaxation properties. On the one hand, Ta has the largest atomic size among these elements. On the other hand, the electronegativity of Ta is the lowest while W is the highest, which results in different charge transfer. Based on a Bader charge analysis [33], we find that on average, the charges on each atom species are 12.55, 10.83, 12.04, and 12.58 e-/atom for V, Ta, Cr, and W respectively in the relaxed supercell. The results indicate that Ta loses 0.17 e-/atom while W gains 0.58 e-/atom in the HEA on average. Compared to the values obtained in the unrelaxed supercell, we find that the Ta loses 0.02 e-/atom upon relaxation, while charge transfer on other atoms are very small. The charge transfer results in positively charged Ta and negatively charged W atoms.
Fig. 2. Averaged partial density of states (PDOS) of d orbitals for each element in the HEA. The black line denotes the PDOS in the unrelaxed structure while the red line correspond to PDOS in the relaxed structure.
3.2. Vacancy energetics
The calculated elemental chemical potential for each element in the HEA is provided in Table 2, together with energy per atom obtained from their corresponding pure bulk metals. The results indicate that the chemical potentials of V and W are lower compared to the energy per atom in their bulk phases. These chemical potentials are then used to calculate the defect formation energies.
Table 2
Table 2 Elemental chemical potential of different elements in the HEA (μ), compared with the energy per atom (ε) in their corresponding bulk pure metals.
The calculated formation and migration energies of vacancies are provided in Fig. 3. Similar to defect energetics in other HEAs [10,18], these energies exhibit wide distributions. The formation energies range from 2.45 to 3.94 eV, with an averaged value of 3.18 eV. The energy spread is as large as 1.49 eV, possibly due to the large local lattice distortion. The averaged value is lower than the formation energy in pure W (3.22 eV [34]), but higher than that in pure Ta (2.86 eV [34]). Note that in Ni‒containing fcc HEAs, the calculated formation energies of vacancy are generally higher than that in pure Ni. Besides, the energy spread is less than 1.0 eV [10,18] because of the smaller lattice distortion.
Fig. 3. Formation (a) and migration (b) energy of vacancies in the studied HEA.
We have analyzed the variation of the formation energy with respect to the composition of 1nn shell around the vacancy as given in Fig. 4. Generally, the results indicate that the formation energy increases with increasing V, Cr, and W atoms in the 1nn shell, whereas it decreases with increasing Ta. Inspection of the relaxation distance of all the atoms in the 1nn shell of the removed lattice atom suggests that Ta atoms undergo the largest relaxation, as large as 0.35 Å, as shown in Fig. 5. The averaged relaxation distances for all the vacancies studied are 0.07, 0.16, 0.06, 0.07 Å for V, Ta, Cr, and W respectively. Therefore, the Ta atoms have the largest sizes in this system experience the largest displacement around a vacancy upon relaxation. Surprisingly, the relaxation of W atoms is in the same order as V and Cr, though its size is much larger. The largest relaxation distance of Ta helps to lower the energy of the system, which is in accordance with the decreasing trend of formation energy when the number of Ta atoms in the 1nn shell increases.
Fig. 5. Averaged relaxation distance of different elements in the 1nn shell around a vacancy.
The formation energy of a monovacancy in W-Ta and W-V binary alloys has been calculated previously [32]. It is found that the formation energy in W-Ta alloys strongly depends on the local environment, exhibiting a wide distribution. On the other hand, the distribution in W-V alloys is relatively narrow. Our results show that Ta actually has much larger relaxation around vacancies, leading to the large energy disparity in the W-Ta alloys. The relaxation of W and V is similar, resulting in similar vacancy formation energy in W-V alloys.
The migration energies of vacancies in the HEA range from 0.07 to 1.95 eV. For each vacancy type, at least 32 calculations are performed to sample the variation of the local atomic environment. Based on the calculated results, the averaged migration energies are 1.00, 0.71, 1.11, and 1.15 eV for V, Ta, Cr, and W vacancies, respectively. Thus the migration of a Ta vacancy shows the lowest energy barrier among the four vacancy types. Note that in their corresponding pure bulk metals, the migration barriers of a vacancy are 0.62, 1.48, 0.91, and 1.28 for V, Ta, Cr, and W respectively [29]. From elemental metals to the HEA, the largest difference of vacancy migration barriers is found in Ta; the barrier is the highest among the four elemental metals, but becomes the lowest in the HEA.
In multicomponent HEAs, the formation energy of vacancy is usually elemental specific. In particular, elements with larger atomic sizes tend to exhibit lower vacancy formation energies. Based on our calculated data, the averaged values are 3.20, 3.20, 3.17, 3.13 eV for V, Ta, Cr, and W vacancies respectively. The differences of these values are very small, suggesting no preference of vacancy formation for a specific element. However, the atomic size indeed has a great influence on the migration energies of vacancies. Fig. 3 suggests that the exchange between a Ta atom and a vacancy has the lowest migration barriers compared to others, due to the largest atomic size of Ta. The low migration energy of Ta vacancies indicates that vacancy defects prefer to exchange with nearby Ta atoms, leaving behind Ta-depleted region around vacancy-related defects.
Vacancy-mediated diffusion relates to the phase stability and elemental segregation of materials under irradiation. In the widely studied austenitic FeNiCr alloy, it is found that the oversized solute of Cr generally depletes at grain boundaries, whereas undersized solute of Ni enriches. This phenomenon is a result of the fast diffusion of vacancy flux coupled with Cr [35,36]. In the ion irradiated VTaCrW HEA, a depletion of Ta was also found between the precipitate and the grain matrix [13]. However, W also depletes in the experiment. Our results suggest that the vacancy migration barriers of W are similar to those of Cr and V, indicating that the depletion of W may not related to the vacancy flux only but also depends on other factors such interstitial diffusion.
3.3. Interstitial energetics
In both bcc Ta and W, the [111] dumbbell interstitials have the lowest formation energies [37]. Therefore, we create dumbbell interstitials along the [111] direction to calculate the formation energy of interstitials in the considered WTaCrV alloy. In contrast to fcc HEAs where the added interstitial may be stable approximately at the introduced location, the introduced interstitial undergoes large displacement upon relaxation in bcc HEAs, due to the large atomic size difference. In most cases, the interstitial moves and occupies a lattice position, knocking out other lattice atoms to form interstitial dumbbells. Such defect configuration actually includes an antisite defect and an interstitial dumbbell, which cannot be used to calculate the formation energy of interstitial. We have tried to put the extra atom into many different locations (more than 60 for each interstitial type) and consider only those configurations that can form stable interstitial defects. The calculated formation energies from those refined structures are given in Fig. 6.
Fig. 6. Formation energy of interstitial dumbbells in the V-Ta-Cr-W alloy. Only those stable dumbbells upon relaxation are considered.
Fig. 6 shows that the formation energy of interstitials in the HEA is in the range of 2.08-7.43 eV. This value is much lower than the formation energy in pure W (10.29 eV [34]), but comparable with the formation energies in pure V, Cr, and Ta, which are 2.41, 6.62, and 4.77 eV respectively [34]. In addition, the formation energies of W‒ and Ta‒containing interstitials are larger than those composed of V and Cr, which can be understood by the small atomic size of V and Cr. In fact, lower formation energies of V‒containing interstitial have also been observed in dilute W-V alloys [32]. Among the stable interstitial dumbbells studied, V-V, V-Cr and Cr-Cr exhibit the lowest formation energies. In concentrated alloys, it has been demonstrated that the chemically-biased diffusion is the main mechanism for mass transport under ion irradiation [38,39]. Hence, in the studied HEA, mass transportation will proceed mainly through V and Cr component. Indeed, the irradiated VTaCrW alloys show precipitates enriched by Cr and V [13]. Our analysis based on defect energetics thus is consistent with the experiment results.
Though the initially introduced interstitial dumbbells are along the [111] direction, we find that most dumbbells may change direction or even the bonded lattice atom after energy minimization. For the relaxed interstitial dumbbells, we have analyzed their separation distance and orientation. In accordance with the small atomic size of V and Cr, the separation distance between V—V, V-Cr, and Cr-Cr are smaller than other dumbbells, ranging from 1.90 to 2.10 Å as can be seen from Fig. 7. Inspection of the orientation of all the dumbbells suggests that only 20 % of the dumbbells adopt the [111] direction, whereas the rest convert to the [110] direction, especially for V‒ and Cr‒containing dumbbells. Note that in the pure metals composed of these four elements, the [110] dumbbell is more stable than the [111] ones only in pure Cr [34]. For the other three nonmagnetic pure metals, [111] dumbbells are more stable. Our results thus indicate that HEAs may change this picture; though the HEA is nonmagnetic, the interstitial dumbbells prefer to adopt the [110] direction.
Fig. 7. Separation distance of interstitial dumbbells in the HEA.
The structure of interstitials has a significant implication for their mobilities. It has been found in W that the interstitial moves quickly with a very low energy barrier of 0.05 eV [37]. This is because interstitials adopt [111] orientations in W so that they can easily translate themselves through the lattice along the dumbbell axis direction. Such long-range migration of interstitials has been observed in electron-irradiated W at a low temperature of 4.5 K [40]. However, for interstitials adopt [110] orientations, their migration proceeds through a sequence of rotational and translation jumps [41]. A typical example is an interstitial in Fe, in which interstitial migrates through the three dimensional motion and requires a relatively high activation temperature (100 K) for the diffusion to start [42]. In the HEA, most dumbbells are along the [110] direction, suggesting that the diffusion of interstitials should go through the rotation-translation process, and the overall diffusivity get slowed down. As a result, the defect evolution in the HEA is delayed under irradiation.
4. Discussion
Our calculations have demonstrated that the vacancy migration becomes faster whereas interstitial diffusion becomes slower in the HEA system. This is similar to the results in the widely-studied fcc HEAs, where an overlap region between the migration energy spectrum of vacancies and interstitials is observed [9,10]. Consequently, a large portion of vacancies and interstitials have similar diffusivities, leading to enhanced defect recombination. Notably, in the studied HEA, due to the large lattice distortion, the migration barrier of vacancies can be as low as 0.07 eV, which is comparable to the migration barrier of interstitials even in pure metals. Therefore, much larger overlap region between the migration barriers of vacancies and interstitials is expected in bcc HEAs, contributing to their outstanding irradiation resistance. Indeed, no dislocation loops but only black spots are observed in the studied HEA under ion irradiation up to 8 dpa [13].
In the studied HEA, a strong segregation tendency of Cr and V is evidenced by the calculated negative Warren-Cowley short-range order (SRO) parameter for V-Cr pairs [13]. The result indicates possible phase separations at thermodynamic equilibrium conditions. Under ion irradiation, plenty of defects are created, which may change the segregation tendency through mass transportation mediated by defect flux. The knowledge of defect energetics then can help to analyze the role of defect diffusion on segregation behavior. Based on the results in this study, we predict that interstitial flux is mediated mainly by V and Cr, while vacancies prefer to exchange position with Ta. Therefore, defect flux tends to enrich defect sinks such as grain boundaries with V and Cr. This factor, together with the negative SRO of V-Cr pairs at equilibrium conditions, results in the strong segregation tendency of these two elements under irradiation as observed experimentally [13].
The equiatomic HEA studied in this work is slightly different from the W38Ta36Cr15V11 alloy used in the previous experiment. Here we chose the equiatomic alloy to maximize the chemical disorder so as to obtain unbiased local-environment-dependent defect energetics. The formation energies vary with the changing local atomic environment. Based on the results in Fig. 4, the formation energies of vacancies tend to increase with increasing W, Cr, and V whereas decrease with increasing Ta. The trend is more pronounced for vacancies near Ta. Therefore, in the non-equiatomic W38Ta36Cr15V11, it is expected that the formation energies of vacancies will shift to lower energy directions. For interstitials, they tend to bind with V and Cr. The low concentration of these two elements would lead to a decrease of the portion of such binding, therefore increases the formation energies of interstitials.
The DFT calculations in this work are performed at 0 K. In this case, a wide distribution of migration barriers has been found, assuming random migration path for defects. At finite temperature, defects can find their own optimal migration pathways according to local atomic environment. Based on our calculated migration barriers, vacancies exhibit much lower barriers (as low as 0.07 eV) compared to values in pure metals. As a result, the averaged migration barriers at finite temperature would be lower than the average value at 0 K, since some high-barrier states will not be visited.
The defect energetics is a result of both elemental properties and lattice distortion. In order to distinguish these two contributions, we have calculated the vacancy formation energies in a perfect bcc reference supercell without lattice relaxation. The induced changes in vacancy formation energies are given in Fig. 8. In the un-relaxed supercell, the formation energies are attributed to the constituent elements themselves and their environments, without lattice distortion. Including the lattice distortion by allowing atomic relaxation leads to decrease of vacancy formation energies, especially for V and Cr vacancies. For Ta and W vacancies, there are large fluctuations upon relaxation, suggesting the effect of lattice distortion due to their large atomic volumes. These results indicate that lattice distortion indeed has a large influence on the defect energetics and should be considered when evaluating the defect energetics.
Fig. 8. Difference of vacancy formation energies calculated in the relaxed and un-relaxed supercell.
5. Conclusion
We have presented the structure and defect energetics in a typical HEA with the bcc structure based on DFT calculations. It is found that the local lattice distortion is much larger in the studied HEA due to the large size mismatch among the constituent elements. The atomic size is in the order Ta > W>V > Cr. We show that the formation energies of vacancies only weakly depend on the atomic size of the element. However, the large size of Ta indeed results in much lower migration energies of Ta vacancies compared to others. For interstitials, we show that Ta‒ and W‒containing interstitial dumbbells exhibit lower formation energies than V‒ and Cr‒containing ones. Besides, most interstitial dumbbells favors the [110] direction instead of [111] direction in most nonmagnetic bcc metals. These results, combing vacancy and interstitials energetics, suggest an enrichment of Ta and W and a depletion of V and Cr at defect sinks, which is consistent with available experimental findings. In addition, we show that the spectrum of migration energies for vacancies and interstitials have a much larger overlap region due to the large lattice distortion compared to fcc HEAs studied before, which may contribute to the outstanding irradiation performance of bcc HEA.
Acknowledgment
This work was supported financially by the Project of the City University of Hong Kong (No. 9610425) and the Research Grants Council of Hong Kong (No. 21200919).
It has been shown that concentrated solid solution alloys possess unusual electronic, magnetic, transport, mechanical and radiation-resistant properties that are directly related to underlying chemical complexity. Because every atom experiences a different local atomic environment, the formation and migration energies of vacancies and interstitials in these alloys exhibit a distribution, rather than a single value as in a pure metal or dilute alloy. Using ab initio calculations based on density functional theory and special quasirandom structures, we have characterized the distribution of defect formation energy and migration barrier in four Ni-based solid-solution alloys: Ni0.5Co0.5, Ni0.5Fe0.5, Ni0.8Fe0.2, and Ni0.8Cr0.2. As defect formation energies in finite-size models depend sensitively on the elemental chemical potential, we have developed a computationally efficient method for determining it which takes into account the global composition and the local short-range order. In addition we have compared the results of our ab initio calculations to those obtained from available embedded atom method (EAM) potentials. Our results indicate that the defect formation and migration energies are closely related to the specific atoms in the structure, which further determines the elemental diffusion properties. Different EAM potentials yield different features of defect energetics in concentrated alloys, pointing to the need for additional potential development efforts in order to allow spatial and temporal scale-up of defect and simulations, beyond those accessible to ab initio methods.
xTaTiVCr high-entropy alloy with 32at.% of tungsten (W) and its derivative alloys with 42 to 90at.% of W with in-situ TiC were prepared via the mixing of elemental W, Ta, Ti, V and Cr powders followed by spark plasma sintering for the development of reduced-activation alloys for fusion plasma-facing materials. Characterization of the sintered samples revealed a BCC lattice and a multi-phase structure. The selected-area diffraction patterns confirmed the formation of TiC in the high-entropy alloy and its derivative alloys. It revealed the development of C15 (cubic) Laves phases as well in alloys with 71 to 90at.% W. A mechanical examination of the samples revealed a more than twofold improvement in the hardness and strength due to solid-solution strengthening and dispersion strengthening. This study explored the potential of powder metallurgy processing for the fabrication of a high-entropy alloy and other derived compositions with enhanced hardness and strength.]]>
A body-centered cubic W-based refractory high entropy alloy with outstanding radiation resistance has been developed. The alloy was grown as thin films showing a bimodal grain size distribution in the nanocrystalline and ultrafine regimes and a unique 4-nm lamella-like structure revealed by atom probe tomography (APT). Transmission electron microscopy (TEM) and x-ray diffraction show certain black spots appearing after thermal annealing at elevated temperatures. TEM and APT analysis correlated the black spots with second-phase particles rich in Cr and V. No sign of irradiation-created dislocation loops, even after 8 dpa, was observed. Furthermore, nanomechanical testing shows a large hardness of 14 GPa in the as-deposited samples, with near negligible irradiation hardening. Theoretical modeling combining ab initio and Monte Carlo techniques predicts the formation of Cr- and V-rich second-phase particles and points at equal mobilities of point defects as the origin of the exceptional radiation tolerance.
Two refractory high entropy alloys with compositions near Nb25Mo25Ta25W25 and V20Nb20Mo20Ta20W20, were produced by vacuum arc-melting. Despite containing many constituents, both alloys had a single-phase body-centered cubic (BCC) structure that remained not only stable after exposure to 1400 degrees C, but also disordered, as confirmed by the absence of superlattice reflections in neutron diffraction data. Compressive flow properties and microstructure development of these alloys were determined from room temperature up to 1600 degrees C. Limited compressive plasticity and quasi-cleavage fracture at room temperature suggest that the ductile-to-brittle transition for these alloys occurs above room temperature. At 600 degrees C and above, both alloys showed extensive compressive plastic strain. The yield stress of both alloys dropped by 30-40% between room temperature and 600 degrees C, but was relatively insensitive to temperature above 600 degrees C, comparing favorably with conventional superalloys. (C) 2011 Elsevier Ltd.
Refractory high-entropy alloys (HEAs) are a class of emerging multi-component alloys, showing superior mechanical properties at elevated temperatures and being technologically interesting. However, they are generally brittle at room temperature, fail by cracking at low compressive strains and suffer from limited formability. Here we report a strategy for the fabrication of refractory HEA thin films and small-sized pillars that consist of strongly textured, columnar and nanometre-sized grains. Such HEA pillars exhibit extraordinarily high yield strengths of ∼ 10 GPa--among the highest reported strengths in micro-/nano-pillar compression and one order of magnitude higher than that of its bulk form--and their ductility is considerably improved (compressive plastic strains over 30%). Additionally, we demonstrate that such HEA films show substantially enhanced stability for high-temperature, long-duration conditions (at 1,100 °C for 3 days). Small-scale HEAs combining these properties represent a new class of materials in small-dimension devices potentially for high-stress and high-temperature applications.
A computational method for partitioning a charge density grid into Bader volumes is presented which is efficient, robust, and scales linearly with the number of grid points. The partitioning algorithm follows the steepest ascent paths along the charge density gradient from grid point to grid point until a charge density maximum is reached. In this paper, we describe how accurate off-lattice ascent paths can be represented with respect to the grid points. This improvement maintains the efficient linear scaling of an earlier version of the algorithm, and eliminates a tendency for the Bader surfaces to be aligned along the grid directions. As the algorithm assigns grid points to charge density maxima, subsequent paths are terminated when they reach previously assigned grid points. It is this grid-based approach which gives the algorithm its efficiency, and allows for the analysis of the large grids generated from plane-wave-based density functional theory calculations.
... Tungsten (W) is a leading candidate material as the divertor or the plasma-facing materials (PFMs) in future fusion reactors, owing to its high melting temperature, high thermal conductivity, low sputtering erosion rates, and small tritium retention [[1], [2], [3]]. Despite these advantages, W suffers from its poor mechanical properties due to its low fracture toughness and high brittle to ductile transition temperature (BDTT), which severely restricts its operation temperature window. Besides, irradiation from He ions that can be produced by fusion neutrons may induce significant morphological changes in the surface of W, such as the formation of nanometer-scale bubbles and low-density fuzz structures [2]. These adverse effect has greatly impeded the application of W in fusion reactors. Therefore, various pathways have been proposed and investigated in order to improve the performance of W, mainly focusing on adding minor dopant elements or tailoring its microstructures [4,5]. ...
2
2017
... Tungsten (W) is a leading candidate material as the divertor or the plasma-facing materials (PFMs) in future fusion reactors, owing to its high melting temperature, high thermal conductivity, low sputtering erosion rates, and small tritium retention [[1], [2], [3]]. Despite these advantages, W suffers from its poor mechanical properties due to its low fracture toughness and high brittle to ductile transition temperature (BDTT), which severely restricts its operation temperature window. Besides, irradiation from He ions that can be produced by fusion neutrons may induce significant morphological changes in the surface of W, such as the formation of nanometer-scale bubbles and low-density fuzz structures [2]. These adverse effect has greatly impeded the application of W in fusion reactors. Therefore, various pathways have been proposed and investigated in order to improve the performance of W, mainly focusing on adding minor dopant elements or tailoring its microstructures [4,5]. ...
... ]]. Despite these advantages, W suffers from its poor mechanical properties due to its low fracture toughness and high brittle to ductile transition temperature (BDTT), which severely restricts its operation temperature window. Besides, irradiation from He ions that can be produced by fusion neutrons may induce significant morphological changes in the surface of W, such as the formation of nanometer-scale bubbles and low-density fuzz structures [2]. These adverse effect has greatly impeded the application of W in fusion reactors. Therefore, various pathways have been proposed and investigated in order to improve the performance of W, mainly focusing on adding minor dopant elements or tailoring its microstructures [4,5]. ...
1
2011
... Tungsten (W) is a leading candidate material as the divertor or the plasma-facing materials (PFMs) in future fusion reactors, owing to its high melting temperature, high thermal conductivity, low sputtering erosion rates, and small tritium retention [[1], [2], [3]]. Despite these advantages, W suffers from its poor mechanical properties due to its low fracture toughness and high brittle to ductile transition temperature (BDTT), which severely restricts its operation temperature window. Besides, irradiation from He ions that can be produced by fusion neutrons may induce significant morphological changes in the surface of W, such as the formation of nanometer-scale bubbles and low-density fuzz structures [2]. These adverse effect has greatly impeded the application of W in fusion reactors. Therefore, various pathways have been proposed and investigated in order to improve the performance of W, mainly focusing on adding minor dopant elements or tailoring its microstructures [4,5]. ...
1
2015
... Tungsten (W) is a leading candidate material as the divertor or the plasma-facing materials (PFMs) in future fusion reactors, owing to its high melting temperature, high thermal conductivity, low sputtering erosion rates, and small tritium retention [[1], [2], [3]]. Despite these advantages, W suffers from its poor mechanical properties due to its low fracture toughness and high brittle to ductile transition temperature (BDTT), which severely restricts its operation temperature window. Besides, irradiation from He ions that can be produced by fusion neutrons may induce significant morphological changes in the surface of W, such as the formation of nanometer-scale bubbles and low-density fuzz structures [2]. These adverse effect has greatly impeded the application of W in fusion reactors. Therefore, various pathways have been proposed and investigated in order to improve the performance of W, mainly focusing on adding minor dopant elements or tailoring its microstructures [4,5]. ...
1
2013
... Tungsten (W) is a leading candidate material as the divertor or the plasma-facing materials (PFMs) in future fusion reactors, owing to its high melting temperature, high thermal conductivity, low sputtering erosion rates, and small tritium retention [[1], [2], [3]]. Despite these advantages, W suffers from its poor mechanical properties due to its low fracture toughness and high brittle to ductile transition temperature (BDTT), which severely restricts its operation temperature window. Besides, irradiation from He ions that can be produced by fusion neutrons may induce significant morphological changes in the surface of W, such as the formation of nanometer-scale bubbles and low-density fuzz structures [2]. These adverse effect has greatly impeded the application of W in fusion reactors. Therefore, various pathways have been proposed and investigated in order to improve the performance of W, mainly focusing on adding minor dopant elements or tailoring its microstructures [4,5]. ...
1
2017
... Different from these extensively adopt routes, the development of high-entropy alloy (HEAs) provides another powerful way to tune alloy properties through engineering the chemical disorder in alloys. HEAs are multicomponent alloys composed of different elemental species all at high concentrations [[6], [7], [8]]. Due to the high configurational entropy of mixing, these alloys tend to crystallize as solid solutions in a simple underlying face-centered cubic (fcc) or body-centered cubic (bcc) structures. In HEAs, the local atomic environment of every atom is unique, so that defect properties vary even in the same material. In particular, defect energies, including their formation and migration energies, exhibit distributions [9,10]. As a result, the mutual interaction between vacancies and interstitials may get enhanced, which contributes to the improved defect recombination and accordingly, irradiation resistance [11]. ...
1
2004
... Different from these extensively adopt routes, the development of high-entropy alloy (HEAs) provides another powerful way to tune alloy properties through engineering the chemical disorder in alloys. HEAs are multicomponent alloys composed of different elemental species all at high concentrations [[6], [7], [8]]. Due to the high configurational entropy of mixing, these alloys tend to crystallize as solid solutions in a simple underlying face-centered cubic (fcc) or body-centered cubic (bcc) structures. In HEAs, the local atomic environment of every atom is unique, so that defect properties vary even in the same material. In particular, defect energies, including their formation and migration energies, exhibit distributions [9,10]. As a result, the mutual interaction between vacancies and interstitials may get enhanced, which contributes to the improved defect recombination and accordingly, irradiation resistance [11]. ...
1
2004
... Different from these extensively adopt routes, the development of high-entropy alloy (HEAs) provides another powerful way to tune alloy properties through engineering the chemical disorder in alloys. HEAs are multicomponent alloys composed of different elemental species all at high concentrations [[6], [7], [8]]. Due to the high configurational entropy of mixing, these alloys tend to crystallize as solid solutions in a simple underlying face-centered cubic (fcc) or body-centered cubic (bcc) structures. In HEAs, the local atomic environment of every atom is unique, so that defect properties vary even in the same material. In particular, defect energies, including their formation and migration energies, exhibit distributions [9,10]. As a result, the mutual interaction between vacancies and interstitials may get enhanced, which contributes to the improved defect recombination and accordingly, irradiation resistance [11]. ...
4
2016
... Different from these extensively adopt routes, the development of high-entropy alloy (HEAs) provides another powerful way to tune alloy properties through engineering the chemical disorder in alloys. HEAs are multicomponent alloys composed of different elemental species all at high concentrations [[6], [7], [8]]. Due to the high configurational entropy of mixing, these alloys tend to crystallize as solid solutions in a simple underlying face-centered cubic (fcc) or body-centered cubic (bcc) structures. In HEAs, the local atomic environment of every atom is unique, so that defect properties vary even in the same material. In particular, defect energies, including their formation and migration energies, exhibit distributions [9,10]. As a result, the mutual interaction between vacancies and interstitials may get enhanced, which contributes to the improved defect recombination and accordingly, irradiation resistance [11]. ...
... where Ed and E0 are the energies of defective and defect-free supercells respectively, and μd is the chemical potential of defect species d. The signs of + and - correspond to a vacancy and an interstitial, respectively. In this work, the elemental chemical potentials were estimated following our previous method [9,10,28]. ...
... The distributions of the first and second nearest neighbor (nn) distances are given in Fig. 1(a) and (b). We also calculate the radial displacement of all the atoms as shown in Fig. 1(c), using the perfect bcc lattice as a reference. Consistent with the trend of atomic volumes, the 1nn distance between V and Cr are smaller than those between W and Ta. The distribution of 1nn and 2nn distance shows a slightly overlap region, suggesting the large local lattice distortion effect. The calculated 1nn distances are larger than those reported in fcc HEAs [9]. According to a previous analysis of lattice distortion, the α2 parameter for the studied HEA is 0.04, which is higher than most values of the solid solutions reported in the same paper [31]. This is also an indication of the large lattice distortion in the studied HEA. On the other hand, the radial deviation distance does not exactly follow the trend of atomic size. For example, the larger W exhibits a smaller deviation than the smaller V. The average atomic displacements are 0.094, 0.094, 0.079, and 0.077 Å for V, Ta, Cr, and W respectively. The smaller relaxation displacement of W in the pristine structure is also found in the defected structures, as detailed in the next section. ...
... Our calculations have demonstrated that the vacancy migration becomes faster whereas interstitial diffusion becomes slower in the HEA system. This is similar to the results in the widely-studied fcc HEAs, where an overlap region between the migration energy spectrum of vacancies and interstitials is observed [9,10]. Consequently, a large portion of vacancies and interstitials have similar diffusivities, leading to enhanced defect recombination. Notably, in the studied HEA, due to the large lattice distortion, the migration barrier of vacancies can be as low as 0.07 eV, which is comparable to the migration barrier of interstitials even in pure metals. Therefore, much larger overlap region between the migration barriers of vacancies and interstitials is expected in bcc HEAs, contributing to their outstanding irradiation resistance. Indeed, no dislocation loops but only black spots are observed in the studied HEA under ion irradiation up to 8 dpa [13]. ...
6
2018
... Different from these extensively adopt routes, the development of high-entropy alloy (HEAs) provides another powerful way to tune alloy properties through engineering the chemical disorder in alloys. HEAs are multicomponent alloys composed of different elemental species all at high concentrations [[6], [7], [8]]. Due to the high configurational entropy of mixing, these alloys tend to crystallize as solid solutions in a simple underlying face-centered cubic (fcc) or body-centered cubic (bcc) structures. In HEAs, the local atomic environment of every atom is unique, so that defect properties vary even in the same material. In particular, defect energies, including their formation and migration energies, exhibit distributions [9,10]. As a result, the mutual interaction between vacancies and interstitials may get enhanced, which contributes to the improved defect recombination and accordingly, irradiation resistance [11]. ...
... where Ed and E0 are the energies of defective and defect-free supercells respectively, and μd is the chemical potential of defect species d. The signs of + and - correspond to a vacancy and an interstitial, respectively. In this work, the elemental chemical potentials were estimated following our previous method [9,10,28]. ...
... The migration barrier of defects is calculated by directly optimizing the saddle configurations, as explained in our previous studies [10]. Different from fcc HEAs investigated previously, the bcc equiatomic WTaCrV alloy considered here exhibit rather large lattice distortions (more details given below). Consequently, some types of defects are not stable upon relaxation at some locations, especially for interstitial defects. Due to this fact, the saddle configuration can barely be found for interstitials, and only the migration barriers of vacancies are considered. In this case, the saddle structure contains a lattice atom locating in the middle of two nearest vacancies. By fixing the position of this atom, we calculate the energy of this saddle structure as Es, and the barrier can then be obtained from Em=Es-Ed, where Ed is the energy of the system containing the initial vacancy. ...
... The calculated formation and migration energies of vacancies are provided in Fig. 3. Similar to defect energetics in other HEAs [10,18], these energies exhibit wide distributions. The formation energies range from 2.45 to 3.94 eV, with an averaged value of 3.18 eV. The energy spread is as large as 1.49 eV, possibly due to the large local lattice distortion. The averaged value is lower than the formation energy in pure W (3.22 eV [34]), but higher than that in pure Ta (2.86 eV [34]). Note that in Ni‒containing fcc HEAs, the calculated formation energies of vacancy are generally higher than that in pure Ni. Besides, the energy spread is less than 1.0 eV [10,18] because of the smaller lattice distortion. ...
... ]). Note that in Ni‒containing fcc HEAs, the calculated formation energies of vacancy are generally higher than that in pure Ni. Besides, the energy spread is less than 1.0 eV [10,18] because of the smaller lattice distortion. ...
... Our calculations have demonstrated that the vacancy migration becomes faster whereas interstitial diffusion becomes slower in the HEA system. This is similar to the results in the widely-studied fcc HEAs, where an overlap region between the migration energy spectrum of vacancies and interstitials is observed [9,10]. Consequently, a large portion of vacancies and interstitials have similar diffusivities, leading to enhanced defect recombination. Notably, in the studied HEA, due to the large lattice distortion, the migration barrier of vacancies can be as low as 0.07 eV, which is comparable to the migration barrier of interstitials even in pure metals. Therefore, much larger overlap region between the migration barriers of vacancies and interstitials is expected in bcc HEAs, contributing to their outstanding irradiation resistance. Indeed, no dislocation loops but only black spots are observed in the studied HEA under ion irradiation up to 8 dpa [13]. ...
1
2019
... Different from these extensively adopt routes, the development of high-entropy alloy (HEAs) provides another powerful way to tune alloy properties through engineering the chemical disorder in alloys. HEAs are multicomponent alloys composed of different elemental species all at high concentrations [[6], [7], [8]]. Due to the high configurational entropy of mixing, these alloys tend to crystallize as solid solutions in a simple underlying face-centered cubic (fcc) or body-centered cubic (bcc) structures. In HEAs, the local atomic environment of every atom is unique, so that defect properties vary even in the same material. In particular, defect energies, including their formation and migration energies, exhibit distributions [9,10]. As a result, the mutual interaction between vacancies and interstitials may get enhanced, which contributes to the improved defect recombination and accordingly, irradiation resistance [11]. ...
1
2017
... Indeed, recent experimental results have demonstrated outstanding radiation resistance in W-based HEAs [12,13]. For example, the W38Ta36Cr15V11 alloy with the bcc structure shows no sign of irradiation-created dislocation loops after ion irradiation up to a dose of 8 displacement per atom (dpa) [13]. The good radiation resistance of the V-Ta-Cr-W alloy is explained by the formation of Cr- and V-rich second-phase precipitate as well as the equal mobility of point defects which favors defect recombination. Besides the promising irradiation performance, it is reported that W-based bcc HEAs exhibit superior mechanical properties at high temperatures compared to Ni-based superalloys [14]. By tuning the fabrication strategy, both yield strength and ductility of these bcc HEAs can be improved [15], making them potential candidate materials for future nuclear applications. More importantly, previous experiments indicate that bcc HEAs almost exhibit no radiation hardening effect [13,16], in contrast to the noticeable radiation hardening observed in previous fcc HEAs. These findings closely relate to the specific distribution of defects generated by energetic ion bombardment. Therefore, it is of paramount significance to evaluate the defect properties in bcc HEAs. Most previous studies about defect properties are focused on HEAs with the fcc structures [9,10,[17], [18], [19]], and their properties in bcc HEAs still remain unexplored. ...
8
2019
... Indeed, recent experimental results have demonstrated outstanding radiation resistance in W-based HEAs [12,13]. For example, the W38Ta36Cr15V11 alloy with the bcc structure shows no sign of irradiation-created dislocation loops after ion irradiation up to a dose of 8 displacement per atom (dpa) [13]. The good radiation resistance of the V-Ta-Cr-W alloy is explained by the formation of Cr- and V-rich second-phase precipitate as well as the equal mobility of point defects which favors defect recombination. Besides the promising irradiation performance, it is reported that W-based bcc HEAs exhibit superior mechanical properties at high temperatures compared to Ni-based superalloys [14]. By tuning the fabrication strategy, both yield strength and ductility of these bcc HEAs can be improved [15], making them potential candidate materials for future nuclear applications. More importantly, previous experiments indicate that bcc HEAs almost exhibit no radiation hardening effect [13,16], in contrast to the noticeable radiation hardening observed in previous fcc HEAs. These findings closely relate to the specific distribution of defects generated by energetic ion bombardment. Therefore, it is of paramount significance to evaluate the defect properties in bcc HEAs. Most previous studies about defect properties are focused on HEAs with the fcc structures [9,10,[17], [18], [19]], and their properties in bcc HEAs still remain unexplored. ...
... alloy with the bcc structure shows no sign of irradiation-created dislocation loops after ion irradiation up to a dose of 8 displacement per atom (dpa) [13]. The good radiation resistance of the V-Ta-Cr-W alloy is explained by the formation of Cr- and V-rich second-phase precipitate as well as the equal mobility of point defects which favors defect recombination. Besides the promising irradiation performance, it is reported that W-based bcc HEAs exhibit superior mechanical properties at high temperatures compared to Ni-based superalloys [14]. By tuning the fabrication strategy, both yield strength and ductility of these bcc HEAs can be improved [15], making them potential candidate materials for future nuclear applications. More importantly, previous experiments indicate that bcc HEAs almost exhibit no radiation hardening effect [13,16], in contrast to the noticeable radiation hardening observed in previous fcc HEAs. These findings closely relate to the specific distribution of defects generated by energetic ion bombardment. Therefore, it is of paramount significance to evaluate the defect properties in bcc HEAs. Most previous studies about defect properties are focused on HEAs with the fcc structures [9,10,[17], [18], [19]], and their properties in bcc HEAs still remain unexplored. ...
... ], making them potential candidate materials for future nuclear applications. More importantly, previous experiments indicate that bcc HEAs almost exhibit no radiation hardening effect [13,16], in contrast to the noticeable radiation hardening observed in previous fcc HEAs. These findings closely relate to the specific distribution of defects generated by energetic ion bombardment. Therefore, it is of paramount significance to evaluate the defect properties in bcc HEAs. Most previous studies about defect properties are focused on HEAs with the fcc structures [9,10,[17], [18], [19]], and their properties in bcc HEAs still remain unexplored. ...
... Vacancy-mediated diffusion relates to the phase stability and elemental segregation of materials under irradiation. In the widely studied austenitic FeNiCr alloy, it is found that the oversized solute of Cr generally depletes at grain boundaries, whereas undersized solute of Ni enriches. This phenomenon is a result of the fast diffusion of vacancy flux coupled with Cr [35,36]. In the ion irradiated VTaCrW HEA, a depletion of Ta was also found between the precipitate and the grain matrix [13]. However, W also depletes in the experiment. Our results suggest that the vacancy migration barriers of W are similar to those of Cr and V, indicating that the depletion of W may not related to the vacancy flux only but also depends on other factors such interstitial diffusion. ...
... Fig. 6 shows that the formation energy of interstitials in the HEA is in the range of 2.08-7.43 eV. This value is much lower than the formation energy in pure W (10.29 eV [34]), but comparable with the formation energies in pure V, Cr, and Ta, which are 2.41, 6.62, and 4.77 eV respectively [34]. In addition, the formation energies of W‒ and Ta‒containing interstitials are larger than those composed of V and Cr, which can be understood by the small atomic size of V and Cr. In fact, lower formation energies of V‒containing interstitial have also been observed in dilute W-V alloys [32]. Among the stable interstitial dumbbells studied, V-V, V-Cr and Cr-Cr exhibit the lowest formation energies. In concentrated alloys, it has been demonstrated that the chemically-biased diffusion is the main mechanism for mass transport under ion irradiation [38,39]. Hence, in the studied HEA, mass transportation will proceed mainly through V and Cr component. Indeed, the irradiated VTaCrW alloys show precipitates enriched by Cr and V [13]. Our analysis based on defect energetics thus is consistent with the experiment results. ...
... Our calculations have demonstrated that the vacancy migration becomes faster whereas interstitial diffusion becomes slower in the HEA system. This is similar to the results in the widely-studied fcc HEAs, where an overlap region between the migration energy spectrum of vacancies and interstitials is observed [9,10]. Consequently, a large portion of vacancies and interstitials have similar diffusivities, leading to enhanced defect recombination. Notably, in the studied HEA, due to the large lattice distortion, the migration barrier of vacancies can be as low as 0.07 eV, which is comparable to the migration barrier of interstitials even in pure metals. Therefore, much larger overlap region between the migration barriers of vacancies and interstitials is expected in bcc HEAs, contributing to their outstanding irradiation resistance. Indeed, no dislocation loops but only black spots are observed in the studied HEA under ion irradiation up to 8 dpa [13]. ...
... In the studied HEA, a strong segregation tendency of Cr and V is evidenced by the calculated negative Warren-Cowley short-range order (SRO) parameter for V-Cr pairs [13]. The result indicates possible phase separations at thermodynamic equilibrium conditions. Under ion irradiation, plenty of defects are created, which may change the segregation tendency through mass transportation mediated by defect flux. The knowledge of defect energetics then can help to analyze the role of defect diffusion on segregation behavior. Based on the results in this study, we predict that interstitial flux is mediated mainly by V and Cr, while vacancies prefer to exchange position with Ta. Therefore, defect flux tends to enrich defect sinks such as grain boundaries with V and Cr. This factor, together with the negative SRO of V-Cr pairs at equilibrium conditions, results in the strong segregation tendency of these two elements under irradiation as observed experimentally [13]. ...
... ]. The result indicates possible phase separations at thermodynamic equilibrium conditions. Under ion irradiation, plenty of defects are created, which may change the segregation tendency through mass transportation mediated by defect flux. The knowledge of defect energetics then can help to analyze the role of defect diffusion on segregation behavior. Based on the results in this study, we predict that interstitial flux is mediated mainly by V and Cr, while vacancies prefer to exchange position with Ta. Therefore, defect flux tends to enrich defect sinks such as grain boundaries with V and Cr. This factor, together with the negative SRO of V-Cr pairs at equilibrium conditions, results in the strong segregation tendency of these two elements under irradiation as observed experimentally [13]. ...
1
2011
... Indeed, recent experimental results have demonstrated outstanding radiation resistance in W-based HEAs [12,13]. For example, the W38Ta36Cr15V11 alloy with the bcc structure shows no sign of irradiation-created dislocation loops after ion irradiation up to a dose of 8 displacement per atom (dpa) [13]. The good radiation resistance of the V-Ta-Cr-W alloy is explained by the formation of Cr- and V-rich second-phase precipitate as well as the equal mobility of point defects which favors defect recombination. Besides the promising irradiation performance, it is reported that W-based bcc HEAs exhibit superior mechanical properties at high temperatures compared to Ni-based superalloys [14]. By tuning the fabrication strategy, both yield strength and ductility of these bcc HEAs can be improved [15], making them potential candidate materials for future nuclear applications. More importantly, previous experiments indicate that bcc HEAs almost exhibit no radiation hardening effect [13,16], in contrast to the noticeable radiation hardening observed in previous fcc HEAs. These findings closely relate to the specific distribution of defects generated by energetic ion bombardment. Therefore, it is of paramount significance to evaluate the defect properties in bcc HEAs. Most previous studies about defect properties are focused on HEAs with the fcc structures [9,10,[17], [18], [19]], and their properties in bcc HEAs still remain unexplored. ...
1
2015
... Indeed, recent experimental results have demonstrated outstanding radiation resistance in W-based HEAs [12,13]. For example, the W38Ta36Cr15V11 alloy with the bcc structure shows no sign of irradiation-created dislocation loops after ion irradiation up to a dose of 8 displacement per atom (dpa) [13]. The good radiation resistance of the V-Ta-Cr-W alloy is explained by the formation of Cr- and V-rich second-phase precipitate as well as the equal mobility of point defects which favors defect recombination. Besides the promising irradiation performance, it is reported that W-based bcc HEAs exhibit superior mechanical properties at high temperatures compared to Ni-based superalloys [14]. By tuning the fabrication strategy, both yield strength and ductility of these bcc HEAs can be improved [15], making them potential candidate materials for future nuclear applications. More importantly, previous experiments indicate that bcc HEAs almost exhibit no radiation hardening effect [13,16], in contrast to the noticeable radiation hardening observed in previous fcc HEAs. These findings closely relate to the specific distribution of defects generated by energetic ion bombardment. Therefore, it is of paramount significance to evaluate the defect properties in bcc HEAs. Most previous studies about defect properties are focused on HEAs with the fcc structures [9,10,[17], [18], [19]], and their properties in bcc HEAs still remain unexplored. ...
1
2019
... Indeed, recent experimental results have demonstrated outstanding radiation resistance in W-based HEAs [12,13]. For example, the W38Ta36Cr15V11 alloy with the bcc structure shows no sign of irradiation-created dislocation loops after ion irradiation up to a dose of 8 displacement per atom (dpa) [13]. The good radiation resistance of the V-Ta-Cr-W alloy is explained by the formation of Cr- and V-rich second-phase precipitate as well as the equal mobility of point defects which favors defect recombination. Besides the promising irradiation performance, it is reported that W-based bcc HEAs exhibit superior mechanical properties at high temperatures compared to Ni-based superalloys [14]. By tuning the fabrication strategy, both yield strength and ductility of these bcc HEAs can be improved [15], making them potential candidate materials for future nuclear applications. More importantly, previous experiments indicate that bcc HEAs almost exhibit no radiation hardening effect [13,16], in contrast to the noticeable radiation hardening observed in previous fcc HEAs. These findings closely relate to the specific distribution of defects generated by energetic ion bombardment. Therefore, it is of paramount significance to evaluate the defect properties in bcc HEAs. Most previous studies about defect properties are focused on HEAs with the fcc structures [9,10,[17], [18], [19]], and their properties in bcc HEAs still remain unexplored. ...
1
2018
... Indeed, recent experimental results have demonstrated outstanding radiation resistance in W-based HEAs [12,13]. For example, the W38Ta36Cr15V11 alloy with the bcc structure shows no sign of irradiation-created dislocation loops after ion irradiation up to a dose of 8 displacement per atom (dpa) [13]. The good radiation resistance of the V-Ta-Cr-W alloy is explained by the formation of Cr- and V-rich second-phase precipitate as well as the equal mobility of point defects which favors defect recombination. Besides the promising irradiation performance, it is reported that W-based bcc HEAs exhibit superior mechanical properties at high temperatures compared to Ni-based superalloys [14]. By tuning the fabrication strategy, both yield strength and ductility of these bcc HEAs can be improved [15], making them potential candidate materials for future nuclear applications. More importantly, previous experiments indicate that bcc HEAs almost exhibit no radiation hardening effect [13,16], in contrast to the noticeable radiation hardening observed in previous fcc HEAs. These findings closely relate to the specific distribution of defects generated by energetic ion bombardment. Therefore, it is of paramount significance to evaluate the defect properties in bcc HEAs. Most previous studies about defect properties are focused on HEAs with the fcc structures [9,10,[17], [18], [19]], and their properties in bcc HEAs still remain unexplored. ...
3
2019
... Indeed, recent experimental results have demonstrated outstanding radiation resistance in W-based HEAs [12,13]. For example, the W38Ta36Cr15V11 alloy with the bcc structure shows no sign of irradiation-created dislocation loops after ion irradiation up to a dose of 8 displacement per atom (dpa) [13]. The good radiation resistance of the V-Ta-Cr-W alloy is explained by the formation of Cr- and V-rich second-phase precipitate as well as the equal mobility of point defects which favors defect recombination. Besides the promising irradiation performance, it is reported that W-based bcc HEAs exhibit superior mechanical properties at high temperatures compared to Ni-based superalloys [14]. By tuning the fabrication strategy, both yield strength and ductility of these bcc HEAs can be improved [15], making them potential candidate materials for future nuclear applications. More importantly, previous experiments indicate that bcc HEAs almost exhibit no radiation hardening effect [13,16], in contrast to the noticeable radiation hardening observed in previous fcc HEAs. These findings closely relate to the specific distribution of defects generated by energetic ion bombardment. Therefore, it is of paramount significance to evaluate the defect properties in bcc HEAs. Most previous studies about defect properties are focused on HEAs with the fcc structures [9,10,[17], [18], [19]], and their properties in bcc HEAs still remain unexplored. ...
... The calculated formation and migration energies of vacancies are provided in Fig. 3. Similar to defect energetics in other HEAs [10,18], these energies exhibit wide distributions. The formation energies range from 2.45 to 3.94 eV, with an averaged value of 3.18 eV. The energy spread is as large as 1.49 eV, possibly due to the large local lattice distortion. The averaged value is lower than the formation energy in pure W (3.22 eV [34]), but higher than that in pure Ta (2.86 eV [34]). Note that in Ni‒containing fcc HEAs, the calculated formation energies of vacancy are generally higher than that in pure Ni. Besides, the energy spread is less than 1.0 eV [10,18] because of the smaller lattice distortion. ...
... ,18] because of the smaller lattice distortion. ...
1
2017
... Indeed, recent experimental results have demonstrated outstanding radiation resistance in W-based HEAs [12,13]. For example, the W38Ta36Cr15V11 alloy with the bcc structure shows no sign of irradiation-created dislocation loops after ion irradiation up to a dose of 8 displacement per atom (dpa) [13]. The good radiation resistance of the V-Ta-Cr-W alloy is explained by the formation of Cr- and V-rich second-phase precipitate as well as the equal mobility of point defects which favors defect recombination. Besides the promising irradiation performance, it is reported that W-based bcc HEAs exhibit superior mechanical properties at high temperatures compared to Ni-based superalloys [14]. By tuning the fabrication strategy, both yield strength and ductility of these bcc HEAs can be improved [15], making them potential candidate materials for future nuclear applications. More importantly, previous experiments indicate that bcc HEAs almost exhibit no radiation hardening effect [13,16], in contrast to the noticeable radiation hardening observed in previous fcc HEAs. These findings closely relate to the specific distribution of defects generated by energetic ion bombardment. Therefore, it is of paramount significance to evaluate the defect properties in bcc HEAs. Most previous studies about defect properties are focused on HEAs with the fcc structures [9,10,[17], [18], [19]], and their properties in bcc HEAs still remain unexplored. ...
1
1994
... Ab initio calculations were carried out based on the density functional theory (DFT), using pseudopotentials generated within the projector augmented wave (PAW) approach [20] as implemented in the Vienna ab initio simulation package [21,22]. The exchange-correlation interactions were described by the generalized gradient approximation (GGA) of the Perdew-Burke-Ernzerhof (PBE) form [23]. The Methfessel-Paxton smearing [24] with a smearing width of 0.1 eV was employed to account for partial occupancies of the metallic HEA systems. Periodic boundary conditions and the supercell approach was used for all calculations. The cutoff energy of plane waves was 300 eV. For the defect formation and migration energy calculations, a 4 × 4×4 bcc supercell containing 128 atoms was used and the Brillouin zone was sampled with a 3 × 3×3 k-point mesh based on the Monkhorst-Pack scheme. The convergence threshold for the total energy and atomic force were 10-4 eV and 10-2 eV/A, respectively. The chemical disorder of the HEAs was modelled using special quasirandom structures (SQS) that were constructed by optimization of the Warren-Cowley short range order (SRO) parameters [25,26] using a Monte-Carlo simulated annealing algorithm [27]. The volume of the supercell was fixed during ionic relaxation; the volume was fitted from the calculated energy-volume relations. ...
1
1996
... Ab initio calculations were carried out based on the density functional theory (DFT), using pseudopotentials generated within the projector augmented wave (PAW) approach [20] as implemented in the Vienna ab initio simulation package [21,22]. The exchange-correlation interactions were described by the generalized gradient approximation (GGA) of the Perdew-Burke-Ernzerhof (PBE) form [23]. The Methfessel-Paxton smearing [24] with a smearing width of 0.1 eV was employed to account for partial occupancies of the metallic HEA systems. Periodic boundary conditions and the supercell approach was used for all calculations. The cutoff energy of plane waves was 300 eV. For the defect formation and migration energy calculations, a 4 × 4×4 bcc supercell containing 128 atoms was used and the Brillouin zone was sampled with a 3 × 3×3 k-point mesh based on the Monkhorst-Pack scheme. The convergence threshold for the total energy and atomic force were 10-4 eV and 10-2 eV/A, respectively. The chemical disorder of the HEAs was modelled using special quasirandom structures (SQS) that were constructed by optimization of the Warren-Cowley short range order (SRO) parameters [25,26] using a Monte-Carlo simulated annealing algorithm [27]. The volume of the supercell was fixed during ionic relaxation; the volume was fitted from the calculated energy-volume relations. ...
1
1993
... Ab initio calculations were carried out based on the density functional theory (DFT), using pseudopotentials generated within the projector augmented wave (PAW) approach [20] as implemented in the Vienna ab initio simulation package [21,22]. The exchange-correlation interactions were described by the generalized gradient approximation (GGA) of the Perdew-Burke-Ernzerhof (PBE) form [23]. The Methfessel-Paxton smearing [24] with a smearing width of 0.1 eV was employed to account for partial occupancies of the metallic HEA systems. Periodic boundary conditions and the supercell approach was used for all calculations. The cutoff energy of plane waves was 300 eV. For the defect formation and migration energy calculations, a 4 × 4×4 bcc supercell containing 128 atoms was used and the Brillouin zone was sampled with a 3 × 3×3 k-point mesh based on the Monkhorst-Pack scheme. The convergence threshold for the total energy and atomic force were 10-4 eV and 10-2 eV/A, respectively. The chemical disorder of the HEAs was modelled using special quasirandom structures (SQS) that were constructed by optimization of the Warren-Cowley short range order (SRO) parameters [25,26] using a Monte-Carlo simulated annealing algorithm [27]. The volume of the supercell was fixed during ionic relaxation; the volume was fitted from the calculated energy-volume relations. ...
1
1996
... Ab initio calculations were carried out based on the density functional theory (DFT), using pseudopotentials generated within the projector augmented wave (PAW) approach [20] as implemented in the Vienna ab initio simulation package [21,22]. The exchange-correlation interactions were described by the generalized gradient approximation (GGA) of the Perdew-Burke-Ernzerhof (PBE) form [23]. The Methfessel-Paxton smearing [24] with a smearing width of 0.1 eV was employed to account for partial occupancies of the metallic HEA systems. Periodic boundary conditions and the supercell approach was used for all calculations. The cutoff energy of plane waves was 300 eV. For the defect formation and migration energy calculations, a 4 × 4×4 bcc supercell containing 128 atoms was used and the Brillouin zone was sampled with a 3 × 3×3 k-point mesh based on the Monkhorst-Pack scheme. The convergence threshold for the total energy and atomic force were 10-4 eV and 10-2 eV/A, respectively. The chemical disorder of the HEAs was modelled using special quasirandom structures (SQS) that were constructed by optimization of the Warren-Cowley short range order (SRO) parameters [25,26] using a Monte-Carlo simulated annealing algorithm [27]. The volume of the supercell was fixed during ionic relaxation; the volume was fitted from the calculated energy-volume relations. ...
1
1989
... Ab initio calculations were carried out based on the density functional theory (DFT), using pseudopotentials generated within the projector augmented wave (PAW) approach [20] as implemented in the Vienna ab initio simulation package [21,22]. The exchange-correlation interactions were described by the generalized gradient approximation (GGA) of the Perdew-Burke-Ernzerhof (PBE) form [23]. The Methfessel-Paxton smearing [24] with a smearing width of 0.1 eV was employed to account for partial occupancies of the metallic HEA systems. Periodic boundary conditions and the supercell approach was used for all calculations. The cutoff energy of plane waves was 300 eV. For the defect formation and migration energy calculations, a 4 × 4×4 bcc supercell containing 128 atoms was used and the Brillouin zone was sampled with a 3 × 3×3 k-point mesh based on the Monkhorst-Pack scheme. The convergence threshold for the total energy and atomic force were 10-4 eV and 10-2 eV/A, respectively. The chemical disorder of the HEAs was modelled using special quasirandom structures (SQS) that were constructed by optimization of the Warren-Cowley short range order (SRO) parameters [25,26] using a Monte-Carlo simulated annealing algorithm [27]. The volume of the supercell was fixed during ionic relaxation; the volume was fitted from the calculated energy-volume relations. ...
1
1965
... Ab initio calculations were carried out based on the density functional theory (DFT), using pseudopotentials generated within the projector augmented wave (PAW) approach [20] as implemented in the Vienna ab initio simulation package [21,22]. The exchange-correlation interactions were described by the generalized gradient approximation (GGA) of the Perdew-Burke-Ernzerhof (PBE) form [23]. The Methfessel-Paxton smearing [24] with a smearing width of 0.1 eV was employed to account for partial occupancies of the metallic HEA systems. Periodic boundary conditions and the supercell approach was used for all calculations. The cutoff energy of plane waves was 300 eV. For the defect formation and migration energy calculations, a 4 × 4×4 bcc supercell containing 128 atoms was used and the Brillouin zone was sampled with a 3 × 3×3 k-point mesh based on the Monkhorst-Pack scheme. The convergence threshold for the total energy and atomic force were 10-4 eV and 10-2 eV/A, respectively. The chemical disorder of the HEAs was modelled using special quasirandom structures (SQS) that were constructed by optimization of the Warren-Cowley short range order (SRO) parameters [25,26] using a Monte-Carlo simulated annealing algorithm [27]. The volume of the supercell was fixed during ionic relaxation; the volume was fitted from the calculated energy-volume relations. ...
1
1950
... Ab initio calculations were carried out based on the density functional theory (DFT), using pseudopotentials generated within the projector augmented wave (PAW) approach [20] as implemented in the Vienna ab initio simulation package [21,22]. The exchange-correlation interactions were described by the generalized gradient approximation (GGA) of the Perdew-Burke-Ernzerhof (PBE) form [23]. The Methfessel-Paxton smearing [24] with a smearing width of 0.1 eV was employed to account for partial occupancies of the metallic HEA systems. Periodic boundary conditions and the supercell approach was used for all calculations. The cutoff energy of plane waves was 300 eV. For the defect formation and migration energy calculations, a 4 × 4×4 bcc supercell containing 128 atoms was used and the Brillouin zone was sampled with a 3 × 3×3 k-point mesh based on the Monkhorst-Pack scheme. The convergence threshold for the total energy and atomic force were 10-4 eV and 10-2 eV/A, respectively. The chemical disorder of the HEAs was modelled using special quasirandom structures (SQS) that were constructed by optimization of the Warren-Cowley short range order (SRO) parameters [25,26] using a Monte-Carlo simulated annealing algorithm [27]. The volume of the supercell was fixed during ionic relaxation; the volume was fitted from the calculated energy-volume relations. ...
1
2017
... Ab initio calculations were carried out based on the density functional theory (DFT), using pseudopotentials generated within the projector augmented wave (PAW) approach [20] as implemented in the Vienna ab initio simulation package [21,22]. The exchange-correlation interactions were described by the generalized gradient approximation (GGA) of the Perdew-Burke-Ernzerhof (PBE) form [23]. The Methfessel-Paxton smearing [24] with a smearing width of 0.1 eV was employed to account for partial occupancies of the metallic HEA systems. Periodic boundary conditions and the supercell approach was used for all calculations. The cutoff energy of plane waves was 300 eV. For the defect formation and migration energy calculations, a 4 × 4×4 bcc supercell containing 128 atoms was used and the Brillouin zone was sampled with a 3 × 3×3 k-point mesh based on the Monkhorst-Pack scheme. The convergence threshold for the total energy and atomic force were 10-4 eV and 10-2 eV/A, respectively. The chemical disorder of the HEAs was modelled using special quasirandom structures (SQS) that were constructed by optimization of the Warren-Cowley short range order (SRO) parameters [25,26] using a Monte-Carlo simulated annealing algorithm [27]. The volume of the supercell was fixed during ionic relaxation; the volume was fitted from the calculated energy-volume relations. ...
1
2017
... where Ed and E0 are the energies of defective and defect-free supercells respectively, and μd is the chemical potential of defect species d. The signs of + and - correspond to a vacancy and an interstitial, respectively. In this work, the elemental chemical potentials were estimated following our previous method [9,10,28]. ...
2
2007
... For the VTaCrW alloy, we first calculate its equilibrium lattice constant based on the obtained energy-volume relation. The result is 3.102 Å, which is in the range of the lattice constants of the four corresponding pure constituent metals (3.04 Å for V, 3.31 Å for Ta, 2.85 Å for Cr, and 3.18 Å for W [29]). Based on the calculated result, we calculate the formation enthalpy of this HEA to be 0.11 eV/formula at 0 K. After a full relaxation, it is found that the magnetic moment diminished in this system, even if spin-polarized calculations are performed. Therefore, nonmagnetic calculations were carried out for all the following calculations. ...
... The migration energies of vacancies in the HEA range from 0.07 to 1.95 eV. For each vacancy type, at least 32 calculations are performed to sample the variation of the local atomic environment. Based on the calculated results, the averaged migration energies are 1.00, 0.71, 1.11, and 1.15 eV for V, Ta, Cr, and W vacancies, respectively. Thus the migration of a Ta vacancy shows the lowest energy barrier among the four vacancy types. Note that in their corresponding pure bulk metals, the migration barriers of a vacancy are 0.62, 1.48, 0.91, and 1.28 for V, Ta, Cr, and W respectively [29]. From elemental metals to the HEA, the largest difference of vacancy migration barriers is found in Ta; the barrier is the highest among the four elemental metals, but becomes the lowest in the HEA. ...
1
2009
... From the equilibrium lattice structure, we have studied the atomic displacement of different elements. Based on a Voronoi tessellation analysis [30], the averaged atomic volume for each element can be deduced. The results are provided in Table 1, which shows that the size of these elements is in the order Cr < V<W < Ta. This order is consistent with their atomic volumes in their pure bcc bulk metals. The difference in the atomic volumes is as large as 1 Å3, which is much larger compared to fcc HEAs. ...
1
2015
... The distributions of the first and second nearest neighbor (nn) distances are given in Fig. 1(a) and (b). We also calculate the radial displacement of all the atoms as shown in Fig. 1(c), using the perfect bcc lattice as a reference. Consistent with the trend of atomic volumes, the 1nn distance between V and Cr are smaller than those between W and Ta. The distribution of 1nn and 2nn distance shows a slightly overlap region, suggesting the large local lattice distortion effect. The calculated 1nn distances are larger than those reported in fcc HEAs [9]. According to a previous analysis of lattice distortion, the α2 parameter for the studied HEA is 0.04, which is higher than most values of the solid solutions reported in the same paper [31]. This is also an indication of the large lattice distortion in the studied HEA. On the other hand, the radial deviation distance does not exactly follow the trend of atomic size. For example, the larger W exhibits a smaller deviation than the smaller V. The average atomic displacements are 0.094, 0.094, 0.079, and 0.077 Å for V, Ta, Cr, and W respectively. The smaller relaxation displacement of W in the pristine structure is also found in the defected structures, as detailed in the next section. ...
3
2011
... The different displacement of these elements relates to their different electronic structures. In particular, the studied VTaCrW alloy includes both 3d metals (V and Cr) and 5d metals (Ta and W). It has been shown that the bonding between 3d‒5d metals has stronger covalent character than 5d‒5d metals [32], which affects the relaxation properties of elements. The averaged partial density of states (PDOS) of d orbitals for each element is given in Fig. 2. It shows that 3d Cr and V exhibit a large DOS value at the Fermi energy (N(EF)), compared to 5d Ta and W, an indication of the lattice instability of Cr and V. Indeed, relaxation helps to lower N(EF), thus stabilizing the lattice. This factor may contribute to the large displacement of V and Cr. The Fermi energy locates in the pseudogap of the PDOS, separating the t2g and eg states. Relaxation leads to a shift of the low-energy t2g states toward left, causing a larger separation of the t2g and eg states. The PDOS of Ta and W are very similar. Nevertheless, the displacement of Ta is much larger than W, which suggests that other factors also come to play in determining their relaxation properties. On the one hand, Ta has the largest atomic size among these elements. On the other hand, the electronegativity of Ta is the lowest while W is the highest, which results in different charge transfer. Based on a Bader charge analysis [33], we find that on average, the charges on each atom species are 12.55, 10.83, 12.04, and 12.58 e-/atom for V, Ta, Cr, and W respectively in the relaxed supercell. The results indicate that Ta loses 0.17 e-/atom while W gains 0.58 e-/atom in the HEA on average. Compared to the values obtained in the unrelaxed supercell, we find that the Ta loses 0.02 e-/atom upon relaxation, while charge transfer on other atoms are very small. The charge transfer results in positively charged Ta and negatively charged W atoms. ...
... The formation energy of a monovacancy in W-Ta and W-V binary alloys has been calculated previously [32]. It is found that the formation energy in W-Ta alloys strongly depends on the local environment, exhibiting a wide distribution. On the other hand, the distribution in W-V alloys is relatively narrow. Our results show that Ta actually has much larger relaxation around vacancies, leading to the large energy disparity in the W-Ta alloys. The relaxation of W and V is similar, resulting in similar vacancy formation energy in W-V alloys. ...
... Fig. 6 shows that the formation energy of interstitials in the HEA is in the range of 2.08-7.43 eV. This value is much lower than the formation energy in pure W (10.29 eV [34]), but comparable with the formation energies in pure V, Cr, and Ta, which are 2.41, 6.62, and 4.77 eV respectively [34]. In addition, the formation energies of W‒ and Ta‒containing interstitials are larger than those composed of V and Cr, which can be understood by the small atomic size of V and Cr. In fact, lower formation energies of V‒containing interstitial have also been observed in dilute W-V alloys [32]. Among the stable interstitial dumbbells studied, V-V, V-Cr and Cr-Cr exhibit the lowest formation energies. In concentrated alloys, it has been demonstrated that the chemically-biased diffusion is the main mechanism for mass transport under ion irradiation [38,39]. Hence, in the studied HEA, mass transportation will proceed mainly through V and Cr component. Indeed, the irradiated VTaCrW alloys show precipitates enriched by Cr and V [13]. Our analysis based on defect energetics thus is consistent with the experiment results. ...
1
2009
... The different displacement of these elements relates to their different electronic structures. In particular, the studied VTaCrW alloy includes both 3d metals (V and Cr) and 5d metals (Ta and W). It has been shown that the bonding between 3d‒5d metals has stronger covalent character than 5d‒5d metals [32], which affects the relaxation properties of elements. The averaged partial density of states (PDOS) of d orbitals for each element is given in Fig. 2. It shows that 3d Cr and V exhibit a large DOS value at the Fermi energy (N(EF)), compared to 5d Ta and W, an indication of the lattice instability of Cr and V. Indeed, relaxation helps to lower N(EF), thus stabilizing the lattice. This factor may contribute to the large displacement of V and Cr. The Fermi energy locates in the pseudogap of the PDOS, separating the t2g and eg states. Relaxation leads to a shift of the low-energy t2g states toward left, causing a larger separation of the t2g and eg states. The PDOS of Ta and W are very similar. Nevertheless, the displacement of Ta is much larger than W, which suggests that other factors also come to play in determining their relaxation properties. On the one hand, Ta has the largest atomic size among these elements. On the other hand, the electronegativity of Ta is the lowest while W is the highest, which results in different charge transfer. Based on a Bader charge analysis [33], we find that on average, the charges on each atom species are 12.55, 10.83, 12.04, and 12.58 e-/atom for V, Ta, Cr, and W respectively in the relaxed supercell. The results indicate that Ta loses 0.17 e-/atom while W gains 0.58 e-/atom in the HEA on average. Compared to the values obtained in the unrelaxed supercell, we find that the Ta loses 0.02 e-/atom upon relaxation, while charge transfer on other atoms are very small. The charge transfer results in positively charged Ta and negatively charged W atoms. ...
5
2019
... The calculated formation and migration energies of vacancies are provided in Fig. 3. Similar to defect energetics in other HEAs [10,18], these energies exhibit wide distributions. The formation energies range from 2.45 to 3.94 eV, with an averaged value of 3.18 eV. The energy spread is as large as 1.49 eV, possibly due to the large local lattice distortion. The averaged value is lower than the formation energy in pure W (3.22 eV [34]), but higher than that in pure Ta (2.86 eV [34]). Note that in Ni‒containing fcc HEAs, the calculated formation energies of vacancy are generally higher than that in pure Ni. Besides, the energy spread is less than 1.0 eV [10,18] because of the smaller lattice distortion. ...
... ]), but higher than that in pure Ta (2.86 eV [34]). Note that in Ni‒containing fcc HEAs, the calculated formation energies of vacancy are generally higher than that in pure Ni. Besides, the energy spread is less than 1.0 eV [10,18] because of the smaller lattice distortion. ...
... Fig. 6 shows that the formation energy of interstitials in the HEA is in the range of 2.08-7.43 eV. This value is much lower than the formation energy in pure W (10.29 eV [34]), but comparable with the formation energies in pure V, Cr, and Ta, which are 2.41, 6.62, and 4.77 eV respectively [34]. In addition, the formation energies of W‒ and Ta‒containing interstitials are larger than those composed of V and Cr, which can be understood by the small atomic size of V and Cr. In fact, lower formation energies of V‒containing interstitial have also been observed in dilute W-V alloys [32]. Among the stable interstitial dumbbells studied, V-V, V-Cr and Cr-Cr exhibit the lowest formation energies. In concentrated alloys, it has been demonstrated that the chemically-biased diffusion is the main mechanism for mass transport under ion irradiation [38,39]. Hence, in the studied HEA, mass transportation will proceed mainly through V and Cr component. Indeed, the irradiated VTaCrW alloys show precipitates enriched by Cr and V [13]. Our analysis based on defect energetics thus is consistent with the experiment results. ...
... ]), but comparable with the formation energies in pure V, Cr, and Ta, which are 2.41, 6.62, and 4.77 eV respectively [34]. In addition, the formation energies of W‒ and Ta‒containing interstitials are larger than those composed of V and Cr, which can be understood by the small atomic size of V and Cr. In fact, lower formation energies of V‒containing interstitial have also been observed in dilute W-V alloys [32]. Among the stable interstitial dumbbells studied, V-V, V-Cr and Cr-Cr exhibit the lowest formation energies. In concentrated alloys, it has been demonstrated that the chemically-biased diffusion is the main mechanism for mass transport under ion irradiation [38,39]. Hence, in the studied HEA, mass transportation will proceed mainly through V and Cr component. Indeed, the irradiated VTaCrW alloys show precipitates enriched by Cr and V [13]. Our analysis based on defect energetics thus is consistent with the experiment results. ...
... Though the initially introduced interstitial dumbbells are along the [111] direction, we find that most dumbbells may change direction or even the bonded lattice atom after energy minimization. For the relaxed interstitial dumbbells, we have analyzed their separation distance and orientation. In accordance with the small atomic size of V and Cr, the separation distance between V—V, V-Cr, and Cr-Cr are smaller than other dumbbells, ranging from 1.90 to 2.10 Å as can be seen from Fig. 7. Inspection of the orientation of all the dumbbells suggests that only 20 % of the dumbbells adopt the [111] direction, whereas the rest convert to the [110] direction, especially for V‒ and Cr‒containing dumbbells. Note that in the pure metals composed of these four elements, the [110] dumbbell is more stable than the [111] ones only in pure Cr [34]. For the other three nonmagnetic pure metals, [111] dumbbells are more stable. Our results thus indicate that HEAs may change this picture; though the HEA is nonmagnetic, the interstitial dumbbells prefer to adopt the [110] direction. ...
1
2010
... Vacancy-mediated diffusion relates to the phase stability and elemental segregation of materials under irradiation. In the widely studied austenitic FeNiCr alloy, it is found that the oversized solute of Cr generally depletes at grain boundaries, whereas undersized solute of Ni enriches. This phenomenon is a result of the fast diffusion of vacancy flux coupled with Cr [35,36]. In the ion irradiated VTaCrW HEA, a depletion of Ta was also found between the precipitate and the grain matrix [13]. However, W also depletes in the experiment. Our results suggest that the vacancy migration barriers of W are similar to those of Cr and V, indicating that the depletion of W may not related to the vacancy flux only but also depends on other factors such interstitial diffusion. ...
1
1998
... Vacancy-mediated diffusion relates to the phase stability and elemental segregation of materials under irradiation. In the widely studied austenitic FeNiCr alloy, it is found that the oversized solute of Cr generally depletes at grain boundaries, whereas undersized solute of Ni enriches. This phenomenon is a result of the fast diffusion of vacancy flux coupled with Cr [35,36]. In the ion irradiated VTaCrW HEA, a depletion of Ta was also found between the precipitate and the grain matrix [13]. However, W also depletes in the experiment. Our results suggest that the vacancy migration barriers of W are similar to those of Cr and V, indicating that the depletion of W may not related to the vacancy flux only but also depends on other factors such interstitial diffusion. ...
2
2019
... In both bcc Ta and W, the [111] dumbbell interstitials have the lowest formation energies [37]. Therefore, we create dumbbell interstitials along the [111] direction to calculate the formation energy of interstitials in the considered WTaCrV alloy. In contrast to fcc HEAs where the added interstitial may be stable approximately at the introduced location, the introduced interstitial undergoes large displacement upon relaxation in bcc HEAs, due to the large atomic size difference. In most cases, the interstitial moves and occupies a lattice position, knocking out other lattice atoms to form interstitial dumbbells. Such defect configuration actually includes an antisite defect and an interstitial dumbbell, which cannot be used to calculate the formation energy of interstitial. We have tried to put the extra atom into many different locations (more than 60 for each interstitial type) and consider only those configurations that can form stable interstitial defects. The calculated formation energies from those refined structures are given in Fig. 6. ...
... The structure of interstitials has a significant implication for their mobilities. It has been found in W that the interstitial moves quickly with a very low energy barrier of 0.05 eV [37]. This is because interstitials adopt [111] orientations in W so that they can easily translate themselves through the lattice along the dumbbell axis direction. Such long-range migration of interstitials has been observed in electron-irradiated W at a low temperature of 4.5 K [40]. However, for interstitials adopt [110] orientations, their migration proceeds through a sequence of rotational and translation jumps [41]. A typical example is an interstitial in Fe, in which interstitial migrates through the three dimensional motion and requires a relatively high activation temperature (100 K) for the diffusion to start [42]. In the HEA, most dumbbells are along the [110] direction, suggesting that the diffusion of interstitials should go through the rotation-translation process, and the overall diffusivity get slowed down. As a result, the defect evolution in the HEA is delayed under irradiation. ...
1
2017
... Fig. 6 shows that the formation energy of interstitials in the HEA is in the range of 2.08-7.43 eV. This value is much lower than the formation energy in pure W (10.29 eV [34]), but comparable with the formation energies in pure V, Cr, and Ta, which are 2.41, 6.62, and 4.77 eV respectively [34]. In addition, the formation energies of W‒ and Ta‒containing interstitials are larger than those composed of V and Cr, which can be understood by the small atomic size of V and Cr. In fact, lower formation energies of V‒containing interstitial have also been observed in dilute W-V alloys [32]. Among the stable interstitial dumbbells studied, V-V, V-Cr and Cr-Cr exhibit the lowest formation energies. In concentrated alloys, it has been demonstrated that the chemically-biased diffusion is the main mechanism for mass transport under ion irradiation [38,39]. Hence, in the studied HEA, mass transportation will proceed mainly through V and Cr component. Indeed, the irradiated VTaCrW alloys show precipitates enriched by Cr and V [13]. Our analysis based on defect energetics thus is consistent with the experiment results. ...
1
2019
... Fig. 6 shows that the formation energy of interstitials in the HEA is in the range of 2.08-7.43 eV. This value is much lower than the formation energy in pure W (10.29 eV [34]), but comparable with the formation energies in pure V, Cr, and Ta, which are 2.41, 6.62, and 4.77 eV respectively [34]. In addition, the formation energies of W‒ and Ta‒containing interstitials are larger than those composed of V and Cr, which can be understood by the small atomic size of V and Cr. In fact, lower formation energies of V‒containing interstitial have also been observed in dilute W-V alloys [32]. Among the stable interstitial dumbbells studied, V-V, V-Cr and Cr-Cr exhibit the lowest formation energies. In concentrated alloys, it has been demonstrated that the chemically-biased diffusion is the main mechanism for mass transport under ion irradiation [38,39]. Hence, in the studied HEA, mass transportation will proceed mainly through V and Cr component. Indeed, the irradiated VTaCrW alloys show precipitates enriched by Cr and V [13]. Our analysis based on defect energetics thus is consistent with the experiment results. ...
1
1975
... The structure of interstitials has a significant implication for their mobilities. It has been found in W that the interstitial moves quickly with a very low energy barrier of 0.05 eV [37]. This is because interstitials adopt [111] orientations in W so that they can easily translate themselves through the lattice along the dumbbell axis direction. Such long-range migration of interstitials has been observed in electron-irradiated W at a low temperature of 4.5 K [40]. However, for interstitials adopt [110] orientations, their migration proceeds through a sequence of rotational and translation jumps [41]. A typical example is an interstitial in Fe, in which interstitial migrates through the three dimensional motion and requires a relatively high activation temperature (100 K) for the diffusion to start [42]. In the HEA, most dumbbells are along the [110] direction, suggesting that the diffusion of interstitials should go through the rotation-translation process, and the overall diffusivity get slowed down. As a result, the defect evolution in the HEA is delayed under irradiation. ...
1
1978
... The structure of interstitials has a significant implication for their mobilities. It has been found in W that the interstitial moves quickly with a very low energy barrier of 0.05 eV [37]. This is because interstitials adopt [111] orientations in W so that they can easily translate themselves through the lattice along the dumbbell axis direction. Such long-range migration of interstitials has been observed in electron-irradiated W at a low temperature of 4.5 K [40]. However, for interstitials adopt [110] orientations, their migration proceeds through a sequence of rotational and translation jumps [41]. A typical example is an interstitial in Fe, in which interstitial migrates through the three dimensional motion and requires a relatively high activation temperature (100 K) for the diffusion to start [42]. In the HEA, most dumbbells are along the [110] direction, suggesting that the diffusion of interstitials should go through the rotation-translation process, and the overall diffusivity get slowed down. As a result, the defect evolution in the HEA is delayed under irradiation. ...
1
1991
... The structure of interstitials has a significant implication for their mobilities. It has been found in W that the interstitial moves quickly with a very low energy barrier of 0.05 eV [37]. This is because interstitials adopt [111] orientations in W so that they can easily translate themselves through the lattice along the dumbbell axis direction. Such long-range migration of interstitials has been observed in electron-irradiated W at a low temperature of 4.5 K [40]. However, for interstitials adopt [110] orientations, their migration proceeds through a sequence of rotational and translation jumps [41]. A typical example is an interstitial in Fe, in which interstitial migrates through the three dimensional motion and requires a relatively high activation temperature (100 K) for the diffusion to start [42]. In the HEA, most dumbbells are along the [110] direction, suggesting that the diffusion of interstitials should go through the rotation-translation process, and the overall diffusivity get slowed down. As a result, the defect evolution in the HEA is delayed under irradiation. ...


 WeChat
WeChat
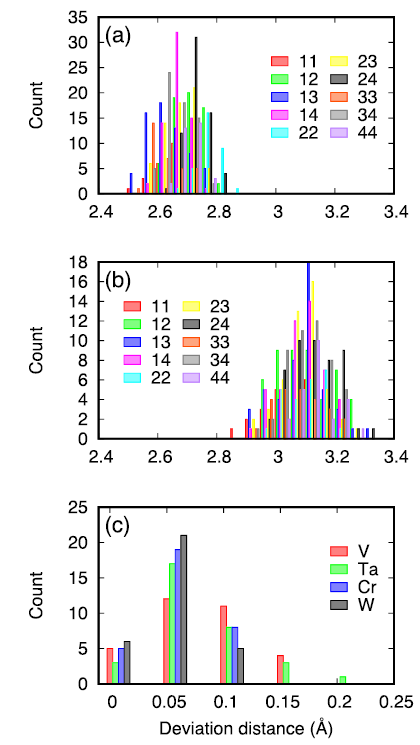

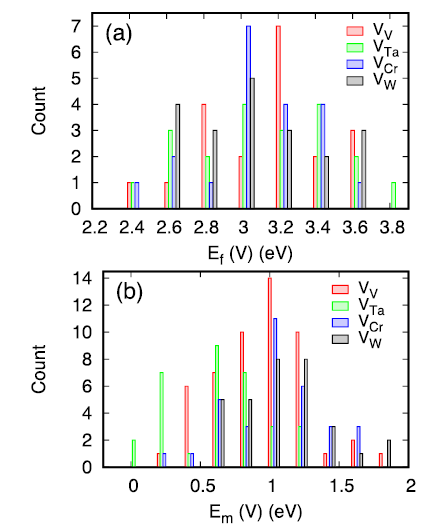
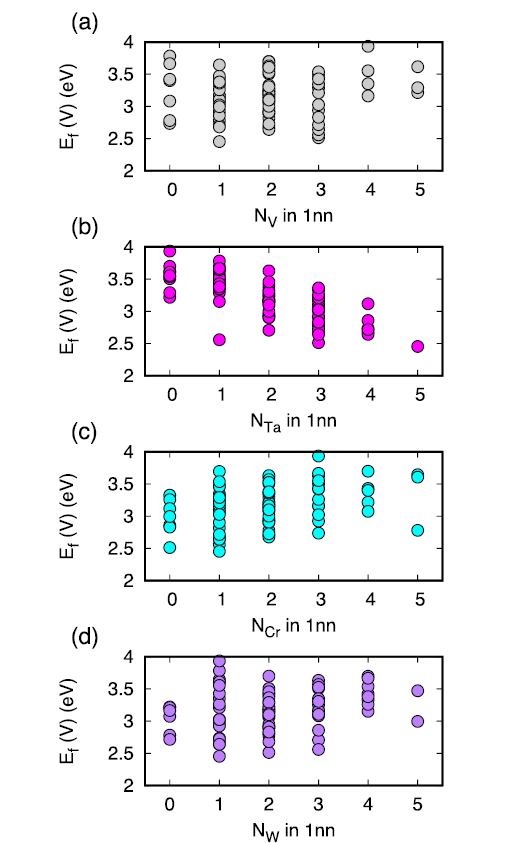
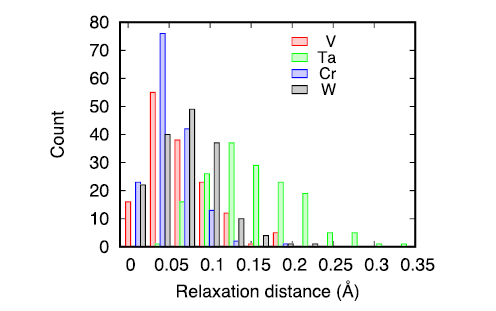
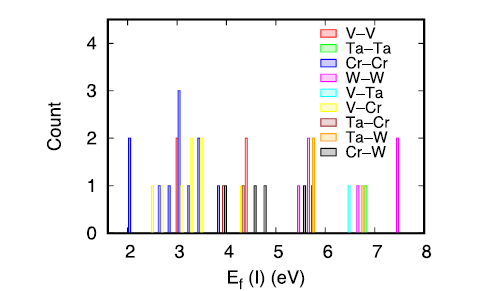
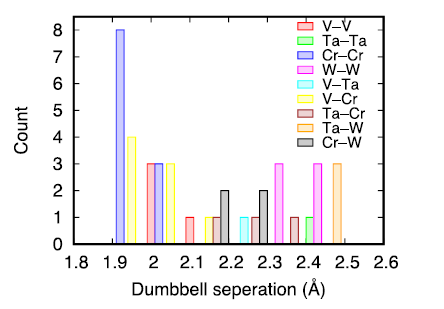
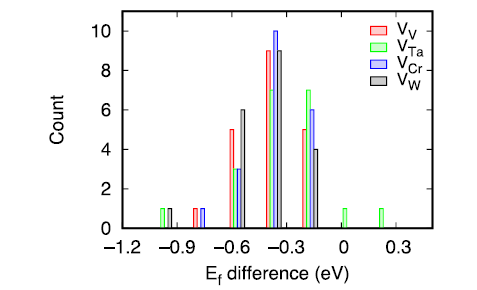
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}