Search for articles:
Fu-Zhi Dai, Bo Wen, Yinjie Sun, Huimin Xiang, Yanchun Zhou
Corresponding authors:
Received: 2019-08-10
Accepted: 2019-09-12
Online: 2020-04-15
Copyright: 2020 Editorial board of Journal of Materials Science & Technology Copyright reserved, Editorial board of Journal of Materials Science & Technology
More
Abstract
High entropy materials (HEMs, e.g. high entropy alloys, high entropy ceramics) have gained increasing interests due to the possibility that they can provide challenge properties unattainable by traditional materials. Though a large number of HEMs have emerged, there is still in lack of theoretical predictions and simulations on HEMs, which is probably caused by the chemical complexity of HEMs. In this work, we demonstrate that the machine learning potentials developed in recent years can overcome the complexity of HEMs, and serve as powerful theoretical tools to simulate HEMs. A deep learning potential (DLP) for high entropy (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C is fitted with the prediction error in energy and force being 9.4 meV/atom and 217 meV/Å, respectively. The reliability and generality of the DLP are affirmed, since it can accurately predict lattice parameters and elastic constants of mono-phase carbides TMC (TM = Ti, Zr, Hf, Nb and Ta). Lattice constants (increase from 4.5707 Å to 4.6727 Å), thermal expansion coefficients (increase from 7.85×10-6 K-1 to 10.58×10-6 K-1), phonon thermal conductivities (decrease from 2.02 W·m-1·K-1 to 0.95 W·m-1·K-1), and elastic properties of high entropy (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C in temperature ranging from 0 °C to 2400 °C are predicted by molecular dynamics simulations. The predicted room temperature properties agree well with experimental measurements, indicating the high accuracy of the DLP. With introducing of machine learning potentials, many problems that are intractable by traditional methods can be handled now. It is hopeful that deep insight into HEMs can be obtained in the future by such powerful methods.
Keywords:
High entropy alloys (HEAs) have attracted great research interests in the last decade, due to the fact that they can display superior mechanical properties, irradiation tolerance, oxidation resistance, and corrosion resistance in comparison to traditional alloys [[1], [2], [3], [4]]. Generally, a typical alloy is composed of one principle metal element with addition of small amount of other elements. In contrast, a HEA is a homogenous solid solute single phase that is composed of four or more principle components in equal or nearly equal molar contents.
It is believed that the multi-principle component state can provide new physical phenomena and challenge properties unattainable by traditional materials. In recent years, the concept of high entropy has been extended to ceramics, where a series of high entropy ceramics (HECs) have emerged, including oxides [5,6], borides [[7], [8], [9]], carbides [[10], [11], [12], [13], [14], [15], [16]], silicides [17], and other inorganic compounds [[18], [19], [20], [21]]. Rost et al. [5] first synthesized a high entropy oxide (Mg0.2Co0.2Ni0.2Cu0.2Zn0.2)O. And later, Bérardan et al. [6] found the high entropy oxide and its derivatives exhibit colossal dielectric constants and superionic conductivity. Gild et al. [7] synthesized a family of high entropy borides, and reported their improved hardness. Sarker et al. [10] systematically evaluated the formation capability of high entropy carbides, measured their lattice parameters, and reported their enhanced hardness. Yan et al. [12] synthesized (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C, and found its low thermal conductivity in nature. Following this, Chen et al. [16] synthesized high porosity high entropy carbides with extremely low thermal conductivity and suggested their possible application in high temperature thermal insulations. In some other high entropy inorganic compounds [21], slower grain growth rate in comparison to traditional compounds has been reported, which is quite relevant to materials applied in high temperatures.
For a deep understanding of high entropy materials (HEMs), fundamental theoretical investigations and computational simulations on HEMs are desired. Due to the chemical complexity and random solid solution state of HEMs, simulations on HEMs are still challenging. To deal with the chemical complexity, methods based on density functional theory (DFT) are the best choice. However, these methods are computational very demanding, which limits their typical simulation length and time scales being nanometers (usually not larger than hundreds of atoms) and picoseconds, respectively. Randomly distributed solid solution state with quite many components cannot be well characterized in such small scales. On the other hand, molecular dynamics (MD) can handle simulations at larger length and longer time scales, which is then suitable to cover the solid solution state with many components. However, MD simulations suffer from lacking of inter-atomic potentials to capture the chemical complexity of high entropy materials, since it is hard to obtain accurate inter-atomic potentials even for solid systems containing three components by traditional methods.
Thanks to the advancement in machine learning and artificial intelligence, new paradigm in atomistic simulations has come forth in the last decade [[22], [23], [24], [25]]. In this new paradigm, machine learning methods are adopted to fit inter-atomic potentials from data sets calculated by DFT methods. The fitted potentials are called machine learning potentials, which bridge the gap between DFT methods and MD simulations. By using machine learning potentials, one can do simulations at cost comparable to MD simulations and with accuracy comparable to DFT methods. Therefore, many problems that are intractable by traditional methods can be handled now. In this work, we fit a machine learning potential for high entropy metal carbide (HEMC) (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C, and use it to predict fundamental thermal and mechanical properties of (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C by MD simulations.
In literatures, many types of machine learning potentials have been published, such as Behler-Parrinello neural network potentials (BPNNPs) [26], Gaussian approximation potentials (GAPs) [27], spectral neighbor analysis potentials (SNAPs) [28] and deep learning potentials (DLPs) [29,30]. The central idea in machine learning potentials is that the energy of a system can be decomposed into energy contributions from constitute atoms, E = ΣiEi (i = 1, 2, …, N). The atomic energy Ei is a function of local environments. Usually, machine learning methods (e.g. neural network, and Gaussian process regression) are used to fit atomic energy with respect to descriptors (also called fingerprints, e.g. symmetry functions, bispectra of neighborhood atomic densities, and smooth overlap of atomic positions SOAP [31,32]) that describe local environment of an atom. For example, in Behler-Parrinello neural network potentials, symmetry functions are adopted as descriptors, and neural networks are used to map descriptors to atomic energies [26].
In most of these machine learning potentials, man designed descriptors are needed for potential training, which involves quite a lot of human intervention. In contrast, DLP utilizes the encoding capability of deep neural networks, and provides an end-to-end potential training framework to minimize the necessary of human intervention. Such an end-to-end potential training framework is particularly relevant to multi-component or multi-phase systems, since we typically have limited knowledge about suitable descriptors for these systems. The potential model adopted in this work was proposed by Zhang et al. [29,30], which is called Deep Potential - Smooth Edition in the original work. For simplicity, the potential model will be called DLP. The DLP consists of two sets of neural networks. One is embedding net, which encodes local environments of atoms automatically to symmetry-preserving descriptors. The other is fitting net, which maps descriptors to atomic energies. For details of the method, please refer to the Refs. [29,30] and the source codes [33,34].
In order to train DLP, a data set should be constructed by using first-principles calculations. Here, first-principles calculations were performed using the Cambridge Serial Total Energy Package (CASTEP) code [35] with the ultrasoft pseudopotential [36] and exchange-correlation described by generalized gradient approximation (GGA) based on the Perdew-Burke-Ernzerhof (PBE) scheme [37]. The plane wave basis set cutoff energy was set to be 400 eV, and k-points mesh with a separation of 0.05 Å-1 according to Monkhorst-Pack method [38] was adopted in the Brillouin zone. 2750 systems were sampled. A system means a randomly distorted cell with its metal elements randomly selected from Ti, Zr, Hf, Nb, Ta. Lattice constant of the cell was selected from 4.2 Å to 4.9 Å to ensure that the smallest and the largest lattice constants of mono-phase carbides were included in the samples. The axes of the cell were chosen to be [111], [11$\bar{2}$]/2 and [1$\bar{1}$0]. Then, the re-orientated cell was distorted in each of its dimensions by a random strain between -3 % and 3 %. For each system, the atoms were first randomly shifted from their previous positions in each direction by a random value selected from a uniform distribution of [-0.4 Å, 0.4 Å], and then the cell was simulated by 10 steps of MD in NVT ensemble with time step of 5 fs and temperature randomly selected between 500 K and 5000 K. Therefore, for each system, 11 samples were obtained with one selected as the test data and others being the training data. Therefore, the data set contains a training set with 27500 samples and a test set with 2750 samples. The architectures of the potential are set as follows: (1) the embedding net includes 3 layers with the nodes in each layer being 60, 120, and 120; (2) the fitting net contains four fully connected layers with 120 nodes in each layer; (3) the projection dimension is selected to be 12; (4) The cutoff distances are set to be (5.8 Å, 6.0 Å).
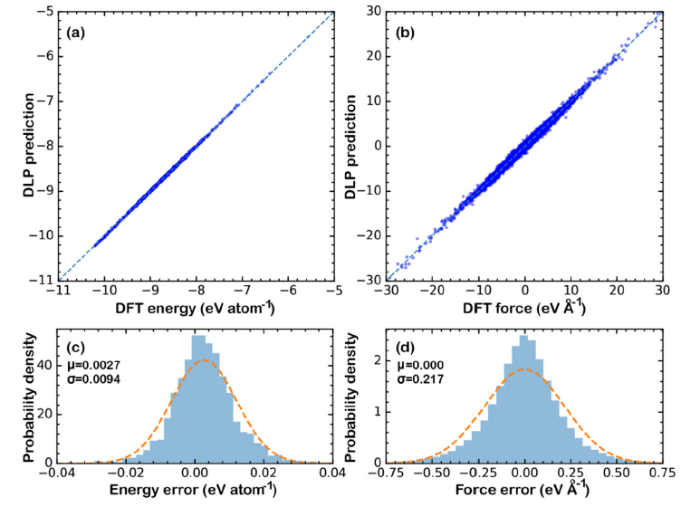
Fig. 1(a) and (b) compares the predicted energies and forces of the test set by DLP with results calculated by DFT method. The figures show that predictions of DLP are highly accurate. The statistics in Fig. 1(c) and (d) show that the prediction errors are normally distributed with standard deviations in energy and force being 9.4 meV/atom and 217 meV/Å, respectively. The error levels are comparable to the results reported by Zhang et al. [30], where they tested the model on HEA with standard deviations in energy and force being 1.68 meV/atom and 394 meV/Å, respectively. To further check the reliability and generality of the fitted DLP, lattice constants and elastic constants of mono-phase carbides were calculated and compared with DFT predictions. The results are shown in Table 1. It can be seen that the DLP accurately predicts lattice constants of mono-phase carbides in comparison with DFT results (results at 0 K). Though the predicted elastic constants display more or less discrepancy to DFT results, the predicted values are still reasonably good. It means that the fitted DLP is highly accurate and has good generalization performance. In the following section, the fitted DLP will be adopted in MD simulations (LAMMPS is used for MD simulations [39]) to predict properties of (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C.
Fig. 1. Comparison of (a) energies and (b) forces predicted by DLP with DFT calculations. Statistical information of prediction error on (c) energies and (d) forces.
Table 1 Comparison of DLP (deep learning potential) predicted lattice constants and elastic constants with DFT results at 0 K.
| Parameter | Potential | TiC | ZrC | HfC | NbC | TaC | HEMC |
|---|---|---|---|---|---|---|---|
| a (Å) | DFT | 4.3315 | 4.7053 | 4.7079 | 4.4802 | 4.5648 | - |
| DLP | 4.3373 | 4.6695 | 4.7011 | 4.4828 | 4.5652 | 4.5669 | |
| c11 (GPa) | DFT | 479 | 477 | 537 | 547 | 549 | - |
| DLP | 473 | 474 | 560 | 552 | 547 | 508 | |
| c12 (GPa) | DFT | 133 | 95 | 100 | 186 | 175 | - |
| DLP | 112 | 116 | 122 | 171 | 213 | 141 | |
| c44 (GPa) | DFT | 154 | 174 | 183 | 136 | 126 | - |
| DLP | 200 | 198 | 201 | 167 | 141 | 184 |
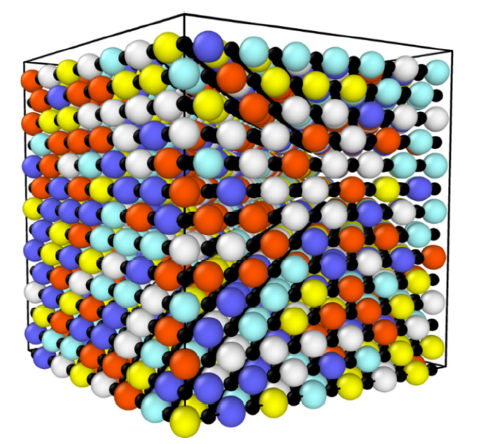
Early transition metal carbides (e.g. ZrC, HfC) crystallize in rock salt structure, which can be viewed as two interpenetrated FCC lattice formed by either metal atoms or carbon atoms. For a HEMC, the metal sites are randomly occupied by different metal elements, as illustrated in Fig. 2. Due to the size differences of these elements, it will result in lattice distortions in HEMs, which is summarized as one of the core effects in HEMs by Yeh et al. [40].
Fig. 2. Schematic illustration of HEMC. Small black atoms are carbon. Colored big atoms are metal atoms. The figure is drawn by using Ovito [
To quantify the lattice distortions in (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C, we take two types of measurements. Firstly, we measured atom deviations along the normal of (111) plane. Taking (111) plane as the reference is because such atomic planes consist of either metal elements or carbon. Namely, we can say that HEMC is formed by alternatively stacking of metal layers and carbon layers along [111] direction. Therefore, deviations of carbon and metal elements can be well separated to characterize fluctuations of the atomic planes. Fig. 3 shows the statistical results of atom deviations from their ideal plane position, which is scaled by interatomic spacing of (111) plane d111. The standard deviations of all atoms, metal atoms and carbon are 0.0129d111, 0.0097d111 and 0.0155d111, respectively. It means that 31 % atoms deviates from their ideal position larger than 1.29 %d111. Inter-layer spacing between carbon and metal atomic planes is 1/2d111, which reveals that mean fluctuations of atomic planes is approaching 2.58 % of inter-atomic plane spacing. In addition, carbon deviates more significantly from the ideal plane positions in comparison with metal elements. The relatively larger deviations of carbon are similar to results reported by Ye at al. [13]. The reason may be that the crystal can be regarded as formed by FCC close packed metal elements with their octahedral interstices occupied by carbon. Therefore, carbon are more freely to deviate from their ideal positions.
Fig. 3. Statistical information on atom deviations from the ideal position of (111) plane (a) all of the atoms, (b) all of the metal atoms and (c) the carbon. The magnitude of deviation is characterized by the interatomic plane spacing of (111) plane, d111.
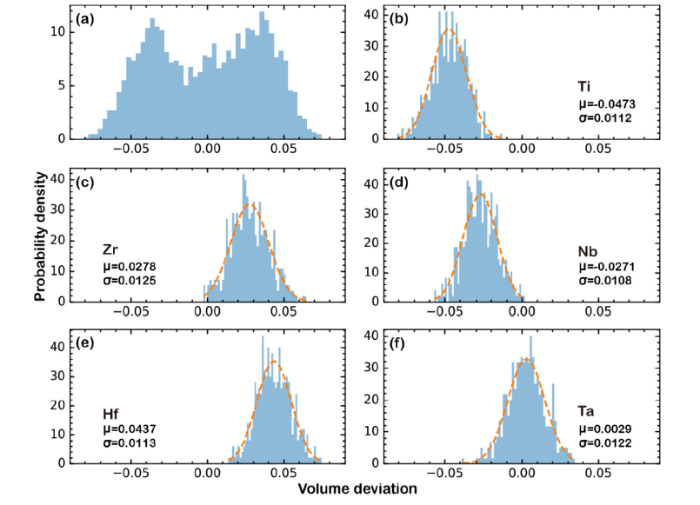
Secondly, we evaluated local volume strains around individual metal atoms. For a given atom, its local elastic strain tensor can be calculated as: εij = (Fij + Fji)/2 - δij, where the deformation gradient matrix F connects a vector in its real strained state and reference unstrained state as: xstrained=Fxunstrained. For each metal atom, xstraineds are the real M-C (M is metal element, C is carbon) bonding vectors, while xunstraineds are unstrained M-C bonding vectors defined by lattice constant of HEMC. Local volume strain is the trace of the elastic strain tensor ε, Σiεii (i = 1, 2, 3). The statistical results of local volume strains are shown in Fig. 4. From the overall results in Fig. 4(a), we can see two evident peaks. One represents local compression strain, while the other is local expansion strain. Fig. 4(b)-(f) shows that Ti and Nb contribute to local compression strains, Zr and Hf contribute to local expansion strains, while volume strain around Ta is almost neutral. It is understandable from lattice constants in Table 1, i.e. lattice constants of TiC and NbC are smaller than that of HEMC, lattice constants of ZrC and HfC are larger than that of HEMC, while lattice constants of TaC and HEMC predicted by the DLP is almost the same.
Fig. 4. Statistical information on local volume strain for (a) all metal atoms, (b) Ti, (c) Zr, (d) Nb, (e) Hf, (f) Ta.
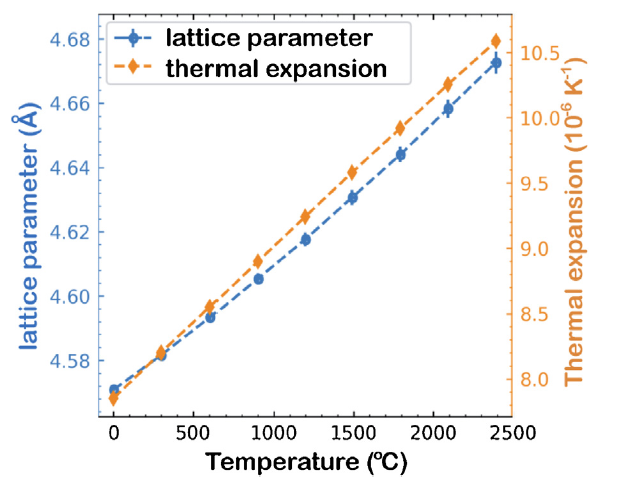
HEMC phases are regarded as promising candidates for applications at ultra-high temperature. It is therefore essential to determine their temperature dependences of properties. Equilibrium lattice constants (a) of (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C at different temperatures were calculated by MD simulations in an NPT ensemble, where the results are plotted in Fig. 5. With temperature increasing from 0 °C to 2400 °C, a increases from 4.5707 Å to 4.6727 Å. The a-T curve can be well fitted by a quadratic function: a = 2.83 × 10-9T2+3.59 × 10-5T+4.5707 with T in °C. Then, thermal expansion coefficients (α) can be determined, which increases linearly from 7.85 × 10-6 K-1 to 10.58 × 10-6 K-1, as shown in Fig. 5. Lattice constant of (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C at room temperature have been measured by Yan et al. [12] (4.518 Å), Zhou et al. [14] (4.5084 Å) and Feng et al. [15] (4.524 Å). As stated by Feng et al. [15], smaller lattice constants in experiments may result from higher oxygen impurities in the sample. Therefore, the lattice constant predicted by the DLP agrees well with experiments. The slightly over estimation in lattice constant of the DLP is probably because the DLP is fitted to GGA DFT data, which usually slightly over estimate lattice parameters. Thermal expansion coefficient predicted by the DLP at room temperature is higher than the measured value 6.44 × 10-6 K-1 by Yan et al. [12]. It reveals that the predicted thermal expansions are probably over-estimated.
Fig. 5. Temperature dependence of lattice parameters and thermal expansion coefficients of HEMC (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C.
Thermal conductivities of (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C were evaluated according to the Green-Kubo method [[42], [43], [44]]. In this method, the thermal conductivity is related to how long it takes the system to dissipate heat flow fluctuations, and the formula is given by: κμν= $\frac{1}{\Omega k_{B}T^{2}} \int^{∞}_{0}\langle J_{\mu}(t) J_{v} (0)\rangle$ dt, where Ω is the system volume, kB is the Boltzmann constant, T is the system temperature, and the angular brackets denote time average of heat current auto-correlation function. Heat current data were collected during simulations in NVT ensemble. For each simulation, the total simulation steps were 5 × 105, data were collected every 10 steps. The inset in Fig. 6 shows a typical correlation function, integration of the correlation function and the running average of the integration. An auto-correlation function reduces fast and converges to zero with a high frequency oscillation around zero, as can be seen in Fig. 6. The decrease in the magnitude of oscillation is very slow, which leads to the necessary of long simulation time. The oscillation in auto-correlation function will result in significant vibration in the integration, which makes the determination of κ not easy. However, the running average of integration converges fast to a stable value, which passes through the middle of the noisy raw data of integration. Therefore, the stable running average is adopted as phonon thermal conductivity κ. The predicted phonon thermal conductivities (κ) are shown in Fig. 6. It reveals that high entropy (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C displays very low phonon thermal conductivities, which decrease slightly from 2.02 W∙m-1∙K-1 to 0.95 W∙m-1∙K-1 in the temperature range of 0 °C-2400 °C. Yan et al. have reported [12] that thermal conductivity of (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C at room temperature is 6.45 W∙m-1∙K-1. The predicted low thermal conductivity agrees well with experiments. Since (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C is electric conductive, the measured data includes contributions from both phonons and electrons. Here, the simulated thermal conductivities contain only phonon contributions. Then, the calculated value is lower than experimental result. The low thermal conductivities from phonons and electrons result from the randomly distributed multi-principle component state of HEMs and the local distortions caused by it, which confines the mean free path of phonons and electrons to be at the scale of lattice constants.
Fig. 6. Temperature dependence of phonon thermal conductivity κ of HEMC (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C. The inset shows a typical correlation function, integration of correlation function and the running average of the integration, which is used to determine thermal conductivity κ.
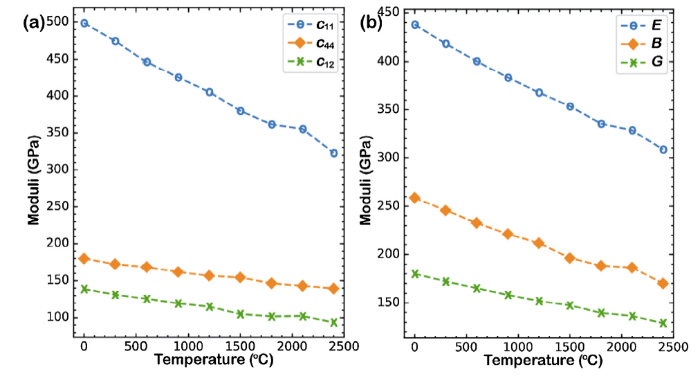
Elastic properties of (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C at different temperatures were calculated by MD simulations in NVT ensembles. A series of deformations with magnitude of ±2 % were applied to the equilibrium crystals, and the resulting stress components were calculated. Then, elastic constants were calculated based on the stress-strain relations. Since (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C is in cubic symmetry, there are three independent elastic constants c11, c44, c12. The temperature dependences of c11, c44, c12 are shown in Fig. 7(a). After elastic constants of the single crystal are obtained, the bulk modulus B and shear modulus G of polycrystalline can be evaluated by applying the Voigt-Reuss-Hill approximations [[45], [46], [47]]. The Voigt approximation and the Reuss approximation for cubic systems are: BV = (c11 + 2c12)/3, GV = (c11 - c12 + 3c44)/5, and BR = 1/(3s11 + 6s12), GR = 5/(4s11 - 4s12 + 3s44), where s11 = (c11 + c12)/(c11×c11 + c11×c12 - c12×c12), s12 = c12/(c12×c12 - c11×c11 - c11×c12), s44 = 1/c44. Voigt approximation and Reuss approximation represent upper and lower bounds for B and G. Hill [47] suggested to use their arithmetic means to estimate B and G: i.e. BH = (BV + BR)/2, GH = (GV + GR)/2. Young’s modulus E can be calculated based on E = 9BG/(3B+G). The variations of B, G, E with respect to temperature are shown in Fig. 7(b). Fig. 7 shows that elastic properties decrease linearly with the increase of temperature (when T > 0 °C), which is consistent with theory. Elastic properties at 0 °C and their decrease speeds with respect to temperature are listed in Table 2. Young’s modulus of (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C at room temperature have been reported by Harrington et al. [11] (∼440 GPa), Yan et al. [12] (479 GPa), and Ye et al. [13] (514-522 GPa) based on nano-indentation experiments. The predicted value by the DLP agrees well with the value reported by Harrington et al. [11], but is lower than other reported values with the largest deviation being less than 20 %. It indicates that the predicted elastic properties by the DLP are reasonably good, since discrepancy in experiments is large. As stated previously, thermal expansion predicted by the DLP may be over-estimated. Thus, elastic properties may be slightly under-estimated by the DLP.
Fig. 7. Temperature dependence of (a) elastic constants (c11, c44, c12) of single crystal HEMC and (b) estimated elastic properties (E, B, G) of polycrystalline HEMC (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C.
Table 2 Values at 0 °C (V0) and decreasing speeds (ΔT) of elastic properties with respect to temperature.
| Parameter | c11 | c44 | c12 | E | B | G |
|---|---|---|---|---|---|---|
| V0 (GPa) | 498 | 178 | 139 | 438 | 259 | 180 |
| ΔT (MPa/oC) | -70.9 | -16.7 | -18.3 | -52.6 | -35.9 | -20.7 |
With the introducing of deep learning potential, we demonstrate that some problems that are intractable by traditional methods can be handled now. The fitted DLP not only predicts high accurate energies and forces in comparison with results calculated by DFT based methods, but also emerges correct properties of mono-phase carbides and high entropy (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C. It indicates that the fitted DLP is suitable for predicting properties of crystalline mono-phase carbides and high entropy (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C. However, it does not mean that it can be applied well for simulating the defect properties of these phases, since no defect state is included in the training data. In this study, we only take the chemical complicity of high entropy materials into account, and any structural complicity (e.g. point defects, dislocations, interfaces, grain boundaries, liquids, etc.) is not considered. In the future, more universal DLP that can accurately characterize both chemical and structural complicities can be fitted for HEMs. Then, it is expected that deep insight into HEMs can be obtained by such powerful methods.
Though machine learning potentials have significantly reduced the computational demanding in comparison to DFT based methods. The typical simulation length and time scales by using machine learning potentials are tens of thousands of atoms and nanoseconds, which is much larger than DFT based methods. However, it is still much smaller than MD simulations with traditional potentials, where millions or even billions of atoms can be practically handled. Therefore, developing machine learning potentials with high accuracy and generality at the minimum computational cost is a meaningful research field, which can further expand the capability of atomistic simulations.
In this study, we fit a DLP for high entropy (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C, which is then used to predict fundamental thermal and mechanical properties of (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C. The main results are summarized below:
(1) The prediction error of DLP in energy and force are 9.4 meV/atom and 217 meV/Å, respectively. The fitted DLP is not only applicable to high entropy (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C, but also predict accurate lattice constants and reasonable elastic constants for mono-phase carbides TMC (TM = Ti, Zr, Hf, Nb and Ta).
(2) Intrinsic lattice distortions of (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C are evaluated, which reveals that the average fluctuations of atomic plane of carbon and metals are 1.55 % and 0.97 %d111 (inter-plane spacing of (111) plane), respectively. The local volume strain states around metal atoms depend on the differences between the lattice constant of (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C and their mono-phase carbides.
(3) Lattice constants (increase from 4.5707 Å to 4.6727 Å), thermal expansion coefficients (increase from 7.85×10-6 K-1 to 10.58×10-6 K-1), phonon thermal conductivities (decrease from 2.02 W·m-1·K-1 to 0.95 W·m-1·K-1), and elastic properties of high entropy (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C in temperature ranging from 0 °C to 2400 °C are predicted, where the room temperature results agree well with experimental measurements.
This work was supported financially by the National Natural Science Foundation of China (Nos. 51672064 and No. U1435206). The authors thank Linfeng Zhang form Princeton University for helpful discussions.

 WeChat
WeChat
/
| 〈 |
|
〉 |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}