Search for articles:
Changjin Xu , Jun Zhou
, Jun Zhou
Corresponding authors:
Received: 2019-07-3
Revised: 2019-08-7
Accepted: 2019-08-22
Online: 2020-03-01
Copyright: 2020 Editorial board of Journal of Materials Science & Technology Copyright reserved, Editorial board of Journal of Materials Science & Technology
More
Abstract
The interface between metal nanoparticles (NPs) and support plays a vital role in catalysis because both electron and atom exchanges occur across the metal-support interface. However, the rational design of interfacial structure facilitating the charge transfer between the neighboring parts remains a challenge. Herein, a guided nucleation strategy based on redox reaction between noble metal precursor and support-surface is introduced to construct epitaxial interfaces between Pt NPs and CeO2 support. The Pt/CeO2 catalyst exhibits near room temperature catalytic activity for CO oxidation that is benefited from the well-defined interface structure facilitating charge transfer from CeO2 support to Pt NPs. Meanwhile, this general approach based on support-surface-induced-nucleation was successfully extended to synthesize Pd and Cu nanocatalysts on CeO2, demonstrating its universal and feasible characteristics. This work is an important step towards developing highly active supported metal catalysts by engineering their interfaces.
Keywords:
Heterogeneous nanocatalysts based on noble metals are widely used in industrial reactions, including energy conversion, pollution control, and producing chemical stocks [[1], [2], [3], [4]]. In order to increase the number of reactive sites accessible for reactant molecules at a reduced cost, nanoparticles (NPs) or clusters of high activity metals are dispersed on porous supports [5,6]. Besides dispersion, the support impact the catalytic performance of metal NPs directly via interfacial interaction. Conventionally, the noble metal nanoparticles are synthesized from the corresponding salts via impregnation/precipitation method that is easy to scale-up for industrial employment [7,8]. However, in most cases, the solution processed metal NPs are physically adhered to the supports and play catalytic role mostly by themselves instead of cooperating with the supports.
The involvement of support in catalyzing reactions goes through the metal-support interfaces and is generally termed as metal-support interaction (MSI) in the literature [9,10]. Correspondingly, the boundary region of the two phases is found to be the virtual source of the exotic catalytic performance of supported nanoparticles. For example, CO oxidation on Au/TiO2 catalysts proceeds at the interface between Au (with adsorbed CO) and TiO2 (with adsorbed O2) [11]. An important step forward in understanding the function of the interface was made by considering the charge transfer [12,13] and intermediate diffusion [14] across the interface. The large electronic perturbation experienced by Pt NPs deposited on CeO2 (111), that does not exist for Pt NPs on the surface of Al2O3 or SiO2, endows Pt the activity to dissociate the O—H bonds in water [12]. The chemical bonding and charge transfer across the interface were then described as electronic metal―support interactions (EMSI) [13]. Coupling the EMSI effect with the rational design of metal NPs and supports opens a new window for developing nanocatalysts with controllable activity and is therefore becoming an active area of interest so far [9,15,16].
Up to date, the major research of tuning the EMSI of nanocatalysts has been concentrated on their components, i.e. composition and structures of the metal nanoparticles and the supports [8,[17], [18], [19], [20], [21], [22]], rather than the quality and nature of the interface itself. For example, the strong EMSI of the Pt-CeO2 catalyst was achieved by enriching the oxygen vacancies in the CeO2 nanorods using a plasma etching treatment [23]. The defective oxygen CeO2 donates electrons to the supported Pt nanoparticles and promotes the catalytic activity for methanol oxidation. A similar effect of improving activity for oxygen reduction reaction (ORR) was observed in Pt NPs loaded on a CeO2-carbon composite [24]. Another strategy to tune the EMSI effect involves morphology control of oxide support—this is effective because both the density of oxygen vacancies [21] and electron transport ability [25] depend on the exposed surface plane. Recently, we have shown that the charge states of nanometals could be precisely controlled by shifting the work function of support [26]. Although the above strategies are definitely effective for enhancing the interfacial interaction, much effort was made to modify the metal NPs or supports rather than directly controlling of the interface itself. The preparation methods are generally complex and non-economical. Generally, the bonding quality of the metal-oxide interface is highly sensitive to the synthesis method. An ideal interface facilitating the electronic perturbation [13] is hardly expected on metal nanoparticles grown in the bulk solution and then attached to the oxide support during evaporation. Hence, it is still an urgent challenge to construct high quality interfaces with strong EMSI effect, thus, to tailor the catalytic properties of metal NPs in a cost-effective way.
In this work, a Pt―CeO2 precursor was first synthesized via a surface redox reaction between Pt ions and trivalent Ce. Followed by the subsequent annealing in H2 flow, the Pt nanoparticles epitaxially grew on the CeO2 support with a coherent interface and a finely tuned electronic interaction (denoted as Pt/CeO2-S). Using CO oxidation as a model reaction, the Pt/CeO2-S nanocatalysts show a strong EMSI effect that tunes the CO adsorption on the Pt sites. This methodology of synthesizing heterocatalysts with the strong electronic metal-support interaction has also been confirmed effective for other transition metals.
Cerium nitrate (Ce(NO3)3·6H2O), chloroplatinic acid (H2PtCI6·6H2O), and sodium hydroxide (NaOH) were used as received from Sinopharm Chemical Reagent Co. Ltd. without any further purification.
The CeO2 nanopowders were obtained by directly calcinating cerium nitrate in a muffle furnace. In brief, a certain amount of cerium nitrate was calcined in air at 500 °C for 5 h.
In a typical procedure, CeO2 nanopowders were first dissolved in 100 ml of deionized water and then ultrasonicated for 30 min. Subsequently, stoichiometric amounts of chloroplatinic acid was added, and the solution was stirred for 2 h to reach adsorption equilibrium. The pH of the above mixed solution was adjusted to ~8 by adding NaOH. The suspension was held at 90 °C for 4 h under vigorous stirring before cooling down to room temperature. The final product was collected by centrifugation, washed with deionized water and ethyl alcohol three times each and then dried at 60 °C in a vacuum oven (DZF-6020, Shanghai Boxun). Finally, the Pt/CeO2-S powders were obtained by heating at 400 °C for 2 h under H2 flow. For comparison, the Pd/CeO2-S and Cu/CeO2-S catalysts were synthesized similarly except that the PdCl2 and Cu(NO3)2·3H2O were used as the respective metal precursors.
For comparison, the sample with the same Pt loading content was also synthesized by a conventional deposition/precipitation method [27]. In brief, a stoichiometric amount of chloroplatinic acid was added to a beaker containing 5 ml of a cerium oxide ethanol solution and stirred for 4 h under vigorous magnetic stirring at room temperature. The pH of this mixed solution was about 3.6. Next, the mixed solution was evaporated completely at 60 °C in a vacuum oven for 12 h. Finally, the Pt/CeO2-C catalyst was obtained by the same calcination procedure as that for Pt/CeO2-S. Furthermore, the Pd/CeO2-C and Cu/CeO2-C were prepared similarly as the Pt/CeO2-C, and the PdCl2 and Cu(NO3)2·3H2O were used as metal precursors.
The XRD patterns were recorded on a X'Pert PRO diffractometer equipped with a CuKα radiation source to reveal crystal structures of catalysts. The actual mass fraction of the Pt in as-prepared catalysts was determined by inductively coupled plasma (ICP) on a Perkin―Elmer Optima 3100 XL spectrometer. The surface areas of the as-synthesized samples (SBET) were determined by Brunauer - Emmett - Teller (BET) method using a Micrometrics Autosorb-TriStar II Apparatus. The TEM images were collected using a JEM-2100 F transmission electron microscope to investigate the dispersion of Pt NPs and the interface between Pt NPs and CeO2 support. The reduction behavior was studied by H2-temperature programmed reduction (TPR). The H2-TPR measurements were carried out in a Micromeritics AutoChem II 2920 Apparatus. The 5.0 % H2-Ar mixed gases were used as the reactive gas stream to pass through 30 mg of the as-prepared catalyst at a flow rate of 50 ml min-1. To investigate the electronic structure of Pt species and the interaction between the support and metal NPs, XPS spectra were recorded on a Perkin Elmer PHI 5000 ESCT System spectrometer with a monochromatic AlKα source where the binding energy values were calibrated using C 1s peak at 284.8 eV. The spectroscopy of FT-IR was collected on a Nicolet 50 FT-IR spectrometer. The CO-TPD experiment was used to study the desorption behavior of CO on Pt sites and was conducted on a U-shaped quartz cuvette. Before the experiments, 30 mg of catalyst was pretreated in a N2 atmosphere for 2 h and then cooled to room temperature. Next, the catalyst was exposed to a 10 % CO/He flow for 1 h followed by N2. Finally, the reaction temperature was increased from room temperature to 400 °C. The composition of the desorbed gases was measured with a mass spectrometer.
To investigate on the changes of adsorption behavior of CO on Pt species under the reaction process, in situ diffuse-reflectance infrared Fourier-transform spectroscopy (DRIFTS) characterization was performed. The in situ DRIFTS characterization was conducted in a Nicolet 50 FT-IR spectrometer using an MCT detector. Before measurement, the catalysts (~ 10 mg) underwent a preprocessing step under N2 atmosphere at 200 °C for 30 min with a flow rate of 40 ml min-1. A background spectrum was collected for spectral correction after cooling to 30 °C. Finally, the catalysts were exposed to mixed gasses (1 % CO, 20 % O2 and N2 as the balance gas) inside the DRIFTS cell. The spectra were averaged over 48 scans with a resolution of 4 cm-1.
The apparent activation energies (Ea) over as-prepared Pt/CeO2 samples were measured under the catalytic reaction test conditions (1% CO, 20% O2, 79% N2) with a space velocity of 24,000 ml·gcat-1 h-1 at different temperatures. The values of Ea were calculated according to the following two equations [28]:
$r=\frac{C_{CO}X_{CO}VP_{atm}}{m_{cat}RT}(mol·s^{-1}·g^{-1}_{cat})$
$lnr=\frac{-E_{a}}{RT}+B$
where r is the reaction rate of as-prepared samples; CCO: molar ratio of CO in mixed gases; XCO: conversion of CO; V: total flow rate of mixed gases (m3·s-1); Patm: atmospheric pressure (1.01 × 105 Pa); mcat: mass of catalyst in the quartz tube reactor (g); R: molar gas constant (8.314 Pa·m3 mol-1 K-1); T: temperature (K); Ea: apparent activation energy (kJ/mol); and B: intercept constant.
The CO oxidation reaction was tested to evaluate the catalytic activity of Pt/CeO2 synthesized via different methods. The reaction was performed on a home-made fixed bed quartz reactor. The 100 mg of catalyst was mixed with an equal volume of quartz sand (Sinopharm Chemical Reagent Co., Ltd) to mix the catalyst evenly. The reaction gas was composed of CO/O2/N2 (1:20:79, volume fraction) with a total flow rate of 40 ml/min. The temperature ramping rate was 10 °C/min. The concentrations of inlet and outlet CO were recorded by an online Agilent 7890A gas chromatograph equipped with a TCD detector.
The CO conversion was defined as:
$X_{CO}=\frac{C_{CO}^{in}-C_{CO}^{out}}{C^{in}_{CO}}\times 100\%$
Where $X_{CO}$ is the CO conversion, $ C_{CO}^{in}$ is the initial concentration of CO, and C_{CO}^{out} is the concentration of CO in the outlet.
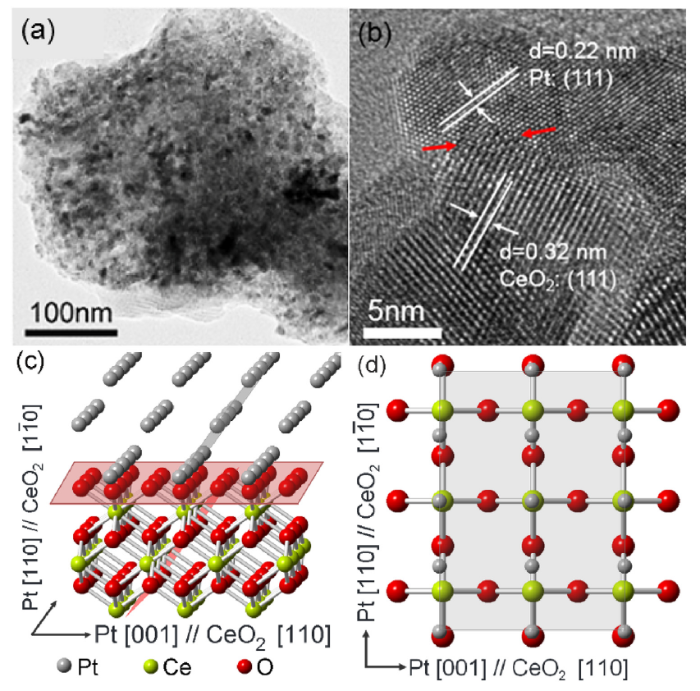
The CeO2 supported Pt NPs were prepared by deposition/precipitation method and subsequent reduction in H2. To build the epitaxial interface, Pt NPs were grown insitu on CeO2via surface redox reaction between Ce3+ and Pt4+. These Pt NPs serve as nuclei for further growth of Pt. TEM images show that the final Pt NPs are homogeneously distributed in the sample prepared by surface redox reaction (Pt/CeO2-S, Fig. 1(a)). For comparison, Pt NPs aggregate on the Pt/CeO2-C and the average size of the Pt NPs are larger than the Pt/CeO2-S catalyst (Fig. S1). HRTEM images show that a sharp interface forms between Pt NPs and CeO2 in the Pt/CeO2-S (Fig. 1(b)). The lattice fringes of 0.32 and 0.22 nm are attributed to the CeO2 (111) and Pt (111), respectively [24,29]. Considering that the measured angles between fringes and the interface (Pt:34°, CeO2:55°), it was deduced that the interface is composed of the Pt (110) plane and the CeO2 (001) under configuration of Pt [001]//CeO2[110], as illustrated in Fig. 1(c,d). The relationship between the Pt and CeO2 is (110) [001]Pt // (001) [110]CeO2, showing perfect epitaxial growth characteristic. The lattice misfits on Pt [001] and its normal direction are less than 4 % and low enough to form coherent interface [30] facilitating the EMSI. For comparison, specific orientation relationship was not found in the Pt/CeO2-C sample.
Fig. 1. TEM (a) and HRTEM (b) images of the Pt/CeO2-S prepared via interfacial nucleation approach. The schematic orientation relation between the Pt NPs and CeO2 support that fits the image of (b): 3D view (c) and top view (d).
The formation of coherent interfaces in the Pt/CeO2-S is mainly driven by the spontaneous redox reaction between Pt4+ and Ce3+ that anchors the initial metallic Pt atomic layer on the CeO2 surface at the nucleation site. The central idea of this work, to construct well-defined Pt―CeO2 interface, is by considering two aspects simultaneously, i.e., promoting the CeO2 surface redox reaction that guides heterogeneous nucleation and suppressing the homogeneous nucleation/growth of Pt NPs (including their precursor precipitates) [31]. Because of the relative low oxygen vacancy formation energy in CeO2 [32], there is a large number of oxygen vacancies (Ov, Fig. S2), creating ample surface Ce3+ species that can reduce the Pt4+. However, the reaction is not fast enough to consume the majority Pt4+. In this work, the redox reaction was accelerated by heating the solution to 90 °C and adjusting the solution pH to weakly alkaline environment (pH ~8). The alkaline environments further favors the surface redox reaction [33]. XPS measurements show that Pt0 species can be only found in Pt/CeO2-S sample before H2 reduction (Fig. S3). In addition, the strong anchoring effect suppresses the agglomeration of Pt NPs during H2 reduction at 400 °C, thus, a Pt―CeO2 interface with chemical bonding of strong interaction forms. In contrast, the growth of Pt NPs in the conventional deposition-precipitation process is confined by the nuclei formation of Pt salt during evaporation condensation [31]. The Pt salt NPs are reduced in the following H2 reduction. In this case, the interface between Pt NPs and CeO2 is formed via simple physical mixing of the two phases, leading to a random interfacial structure and a weaker interaction.
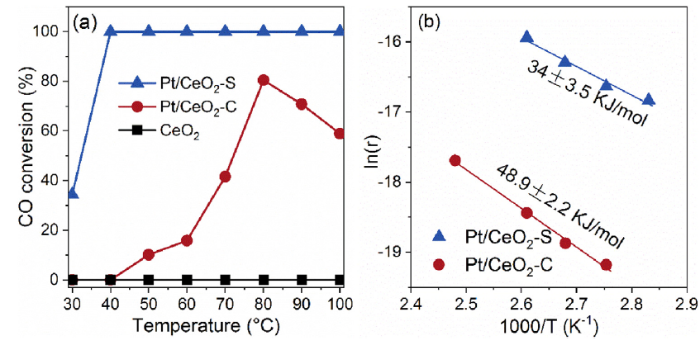
The Pt/CeO2 samples were evaluated by the model reaction of CO oxidation. The CO conversions in Fig. 2(a) demonstrate that the Pt/CeO2-S with coherent interface outperforms the Pt/CeO2-C in a temperature range between 30 and 100 °C. The CO oxidation rate of Pt/CeO2-S catalyst increases rapidly with reaction temperature and reaches complete conversion at 40 °C. In addition, as shown in Fig. 2(b), the apparent activation energy (Ea), measured in the kinetics-control region of Pt/CeO2-S was determined to be 34.0 kJ/mol, which is much lower than the value of Pt/CeO2-C (48.9 kJ/mol). There are likely two contributing factors for the superior catalytic performance of Pt/CeO2-S. First, the fine distribution of Pt NPs size provides more sites for the reaction. Second, the electronic interaction between Pt and CeO2 in Pt/CeO2-S due to the in situ formed coherent interface may be stronger than that in Pt/CeO2-C [34].
Fig. 2. CO conversion over as-prepared Pt/CeO2-S and Pt/CeO2-C catalysts (a) and the activation energy of the Pt/CeO2 catalysts (b).
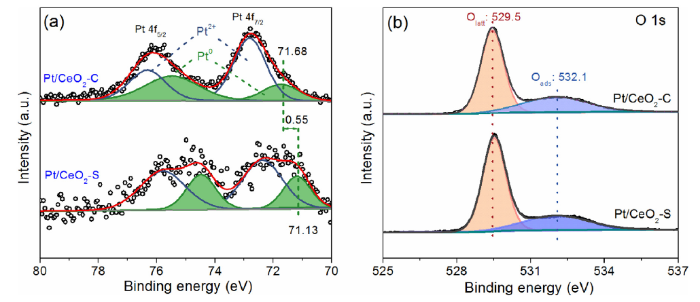
XPS was used to investigate the electronic structure and chemical states of supported Pt nanoparticles prepared by the two methods (Fig. 3). The XPS spectra of the Pt/CeO2-S and Pt/CeO2-C catalysts show typical peaks of Pt 4f7/2 and 4f5/2. Both peaks were deconvoluted into two sub-peaks that correspond to oxidized and metallic Pt. For the Pt/CeO2-C, the two deconvoluted sub-peaks at 71.68 eV and 75.43 eV are attributed to the Pt0 [21,35], while the other two cantered at 72.78 eV and 76.33 eV to the Pt2+ species [36]. The charge states and chemical potential of metal nanoparticles are affected by the oxide support [26,37]. The Pt0 4f binding energy for the Pt/CeO2-S exhibits a downfield shift of ~0.55 eV relative to the Pt/CeO2-C, indicating a higher electronic density of Pt due to charge transfer from CeO2. The charge transfer required by the EMSI is facilitated by the coherent interface between Pt NPs and the CeO2 support. Calculating the relative content of Pt0 based on the areas of fitting peaks (A) by
Pt0%=A(Pt0)/(A(Pt0)+A(Pt2+))×100%
It was found that the Pt/CeO2-S has more metallic Pt0 than the Pt/CeO2-C (Table S1). In addition, the weaker intensities of the Pt 4f signal in Pt/CeO2-S reflect a higher dispersion of Pt [38] that guarantees strong EMSI because of its short-range features [39]. The electron enrichment of Pt downshifts the d-band centre relative to the Fermi level [40]; and this would thus lead to the high reactivity of Pt by donating more electrons to anti-bonding orbitals of the adsorbed intermediates [41], in good agreement with the result of catalytic activity. In short, the strong EMSI is achieved in the Pt/CeO2-S sample due to the well defined interface between Pt NPs and CeO2 support.
Fig. 3. XPS spectra of Pt 4f (a) and O 1s (b) for as-prepared catalysts.
The fine dispersion of Pt in the Pt/CeO2-S sample is achieved mainly because the Pt nucleation is confined by the surface redox reaction initiated by Ce3+. Table S1 presents the content of Ce3+ calculated from the XPS spectra of Ce 3d (Fig. S6). The amount of Ce3+ species is correlated to the density of oxygen vacancies (Ov) [31]. The O 1s XPS spectra in Fig. 3(b) were deconvoluted into two components corresponding to lattice oxygen (Olatt) around 529.5 eV and chemisorbed oxygen (Oads) at 532.1 eV [21]. The two kinds of Pt nanocatalysts have similar amount of oxygen vacancies (about 6 %), suggesting that the consumed Ce3+ due to the surface redox reaction is negligible. CO oxidation over the Pt/CeO2 catalyst is suggested to proceed via the Mars-van Krevelen (M-K) route, which involves chemisorption of CO on Pt and the reaction of lattice oxygen in CeO2 at the Pt―CeO2 interface [35]. The ample oxygen vacancies in CeO2 are vital for improving the catalytic performance.
The results of H2-TPR and CO-TPD were shown in Fig. 4. A temperature-programmed reduction was performed to monitor the reducibility of CeO2, Pt/CeO2-S, and Pt/CeO2-C catalysts with H2 reduction profiles depicted in Fig. 4(a), with the calculated H2 consumption listed in Table S1. The reduction peaks near 330 °C, 462 °C, and 736 °C were seen on CeO2 and originate from the reduction of the surface, sub-surface, and the bulk CeO2, respectively [36]. After Pt loading, an extra reduction peak (I) near 100 °C appears, which is due to reduction of the CeO2 surface neighboring Pt [42]. The reductions of both CeO2 surface (peak I) and CeO2 bulk (peak III) are facilitated at lower temperature because the Pt spills over hydrogen atoms to the CeO2 [43]. In addition, the H2 consumptions near peak I and II for the Pt/CeO2-S are almost doubled than that in the Pt/CeO2-C (see Table S1). Such an increase could be due to the easier hydrogen diffusion promoted by the coherent interface and higher density of oxygen vacancy in the CeO2-S support.
Fig. 4. (a) H2 temperature-programmed reduction curves of CeO2 and Pt/CeO2 catalysts. Peak I: reduction of CeO2 neighboring Pt nanocrystals; peak II: reduction of surface and/or subsurface of CeO2; peak III: reduction of bulk CeO2. (b) CO-TPD patterns of as-synthesized Pt/CeO2 catalysts.
CO-TPD measurements were performed to detect the CO binding because it has a strong influence on catalytic oxidation of CO over Pt [44]. Fig. 4(b) shows three desorption peaks in the CO-TPD profiles. In general, a higher desorption temperature suggests stronger binding of CO. According to the literature, peak (I) is attributed to physical adsorption of CO, and the other two peaks (II and III) at high temperature are due to chemically adsorbed CO in linear and bridge-bonded states, respectively [45]. We note that the desorption temperatures of CO over Pt/CeO2-S is lowered than Pt/CeO2-C, suggesting that the CO chemisorption over Pt/CeO2-S is weakened due to back-donation of electrons into the CO 2π* antibonding orbitals. The weakening of Pt-CO bonding is favourable to suppress CO poisoning effect as seen in previous studies on EMSI effect of Pt supported on WO3 [7] and defect-engineered CeO2 [23]. The adjusting of the CO binding on Pt improves the catalytic activity of CO oxidation near room temperature.
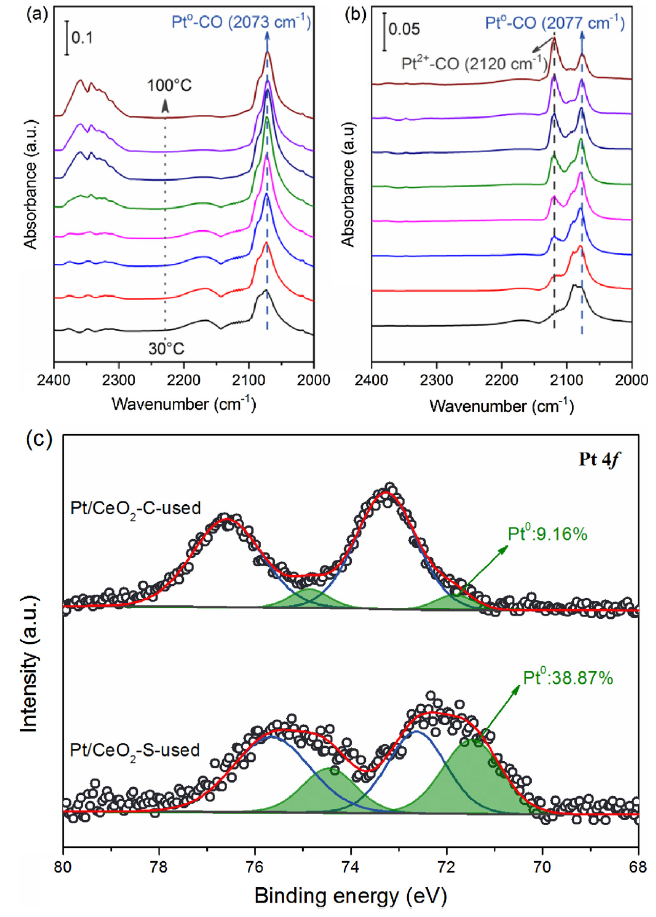
Next, in situ DRIFTS spectra were collected in the kinetics-control region to probe the surface chemistry of Pt/CeO2 catalysts (Fig. 5). As shown in Fig. 5(a), the peak near 2073 cm-1 is attributed to the linear adsorption of CO on Pt0 (denoted as Pt0-CO) [46]. Because the Pt/CeO2-S sample has more Pt0 active sites, the Pt0-CO peak of the Pt/CeO2-S is much stronger in the entire reaction temperature range than that of Pt/CeO2-C, agreeing well with the TEM data. Furthermore, the peak at about 2120 cm-1 is attributed to CO molecules linearly adsorbed on Pt2+ sites (denoted as Pt2+-CO) [47]. This peak for Pt/CeO2-S is weak, and its intensity remains unchanged with increasing reaction temperature. At the same time, the adsorption peaks of gaseous CO2 between 2300-2400 cm-1 emerge and become enhanced during heating, suggesting a progression of CO oxidation. However, the intensity of the Pt2+-CO peak for Pt/CeO2-C gradually increases with temperature indicating that more Pt0 species were oxidized to Pt2+ at higher temperatures. The decay of the Pt chemical state causes decreased catalytic performance for Pt/CeO2-C above 80 °C because the principal active sites for catalytic oxidation of CO are served by metallic Pt0 [48].
Fig. 5. In situ DRIFTS study of CO adsorption over (a) Pt/CeO2-S and (b) Pt/CeO2-C catalysts. (1% CO, 20% O2 and N2 as balance). (c) XPS spectra of the Pt 4f for used Pt/CeO2 catalysts.
The stability of metallic Pt0 during reaction depends on interactions with supports. This is seen in the XPS spectra of Pt 4f after catalysis (Fig. 5(c)). The relative content of Pt0 species (9.16%) in the used Pt/CeO2-C is much lower than that in the fresh Pt/CeO2-C (38.12%) because of the oxidation of Pt during the reaction. However, most of the metallic Pt0 in the Pt/CeO2-S is maintained after the reaction. It is believed that the stronger EMSI in Pt/CeO2-S than in Pt/CeO2-C is responsible for the stability of the Pt0 states in the oxidative environment.
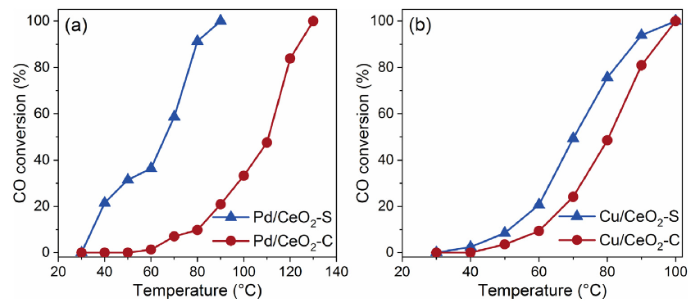
Other metal catalysts (Pd and Cu nanocatalysts) were also prepared via a similar procedure as Pt/CeO2-S to confirm the universal characteristics of the current synthesis method. The CeO2 surface is capable of reducing both Pd and Cu precursors by surface redox reactions. The activity of the resulted Pd/CeO2 and Cu/CeO2 catalysts for CO oxidation were presented on the Fig. 6. For both elements, the catalytic activities of nanocatalysts synthesized by surface redox reaction approach are higher than the corresponding samples prepared by conventional precipitation method. And Pd nanocatalyst exhibits a stronger enhancement effect than Cu, which may result from the different reduction potentials of Pd and Cu precursors. The enhanced catalytic performances demonstrate that the synthetic strategy described here of controlling metal species nucleation during spontaneous redox oxidation reaction is a universal tool for engineering the interface quality of metal-oxide heterostructure nanocatalyst.
Fig. 6. CO conversion over as-prepared Pd/CeO2 (a) and Cu/CeO2 catalysts (b).
In summary, a universal strategy to synthesize Pt/CeO2 nanocatalysts with a coherent interface by a support-surface-nucleation strategy was proposed. This synthesis method takes advantage of surface redox reactions between the Pt precursor and the Ce3+, which confines the subsequent nucleation and growth on the oxide support surface and leads to formation of a coherent interface. Investigations show that the interfacial nucleation strategy favors the dispersion of Pt NPs with smaller particle size leading to more Pt-CeO2 interfaces. A near room temperature activity for CO oxidation is obtained on Pt/CeO2 with well defined interface. The strong EMSI has been confirmed by XPS, in situ DRIFTS, and CO-TPD. The weakened CO adsorption on Pt and activation of CeO2 by Pt collectively promote the catalytic properties of Pt/CeO2. The strategy proposed here is promising in the design of advanced supported metal catalysts for heterogeneous catalysis.
The work was supported by the National Natural Science Foundation of China (Nos.51771047, 51525101, U1602275, 51601119) and the Fundamental Research Funds for the Central Universities (N180204014). J Chen thanks the Key Lab for ATM of Northeastern University (China) and the Natural Science Foundation of Shenzhen University (No. 2019006).
Supplementary material related to this article can be found, inthe online version, at doi: https://doi.org/10.1016/j.jmst.2019.08.036.

 WeChat
WeChat
/
| 〈 |
|
〉 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}