Search for articles:
Meigui Xu , Yujuan Shen, Jun Wang
, Yujuan Shen, Jun Wang
Corresponding authors:
Received: 2019-01-10
Revised: 2019-03-11
Accepted: 2019-03-21
Online: 2020-02-15
Copyright: 2020 Editorial board of Journal of Materials Science & Technology Copyright reserved, Editorial board of Journal of Materials Science & Technology
More
Abstract
The requirement for a sustainable and renewable energy has inspired substantial interests in designing and developing earth-abundant and high-effectiveness electrocatalysts/electrodes for fuel cells and metal-air batteries, in which oxygen reduction reaction (ORR) plays a crucial role. Perovskite oxides have acquired rapid attention as ORR electrocatalysts to replace noble-metal-based catalysts owing to their intrinsic electrocatalytic activity, compositional and structural flexibility. Herein, we report a new Sc and P co-doped perovskite oxide (La0.8Sr0.2Mn0.95Sc0.025P0.025O3-δ, LSMSP) as an active and robust electrocatalyst for the ORR in an alkaline solution. LSMSP electrocatalyst shows superior ORR activity and stability than those of pristine La0.8Sr0.2MnO3-δ (LSM), Sc-doped LSM and P-doped LSM due to the optimized average valence of Mn ions, the large surface area, the smaller particle size and the synergetic effect introduced by the co-doping. Moreover, compared to the benchmark Pt/C electrocatalyst, LSMSP electrocatalyst displays comparable ORR activity and superior durability. These above results suggest that the co-doping strategy of Sc and P into perovskites is a useful method to design high-performance electrocatalysts for the ORR, which can be used in other electrocatalysis-based applications.
Keywords:
Among numerous energy conversion devices, proton exchange membrane fuel cells (PEMFCs) are extremely attractive for automotive applications and portable devices [[1], [2], [3]], in which the electrochemical oxygen reduction reaction (ORR) has been mostly studied [[4], [5], [6], [7], [8], [9], [10], [11]]. However, the sluggish ORR kinetics has been considered as one of the bottlenecks and should be overcome in advancing PEMFC technology. Currently, platinum (Pt) is the state-of-the-art ORR electrocatalyst and shows high electrocatalytic activity [[12], [13], [14]]. Nevertheless, the high price, limited reserves and relatively low stability of Pt catalysts greatly limit its large-scale application [15,16].
The design and development of new electrocatalysts for ORR are mainly categorized into two classes. One is reducing the amount of Pt by constructing alloys [17], composites [18], etc., and the other is replacing Pt with abundant and inexpensive materials [19,20]. Tremendous research advances have been reported in the development of noble-metal-free electrocatalysts for the ORR [[21], [22], [23], [24], [25]], including transition metals [21], metal oxides [21], metal sulfides [22], metal nitrides [23], carbons [24], and boron nitrides [25]. Among them, metal oxides have attracted increasing interests because of their high electrocatalytic activity, superior durability, abundance and low cost [[26], [27], [28]].
As one important class of metal oxides, perovskite oxides with a general formula of ABO3 have shown considerable ORR activity at room temperature, which can be further improved by tuning the compositional elements since 90% of the elements in Periodic Table of Elements can be incorporated into the perovskite structure [[29], [30], [31], [32]]. La1-xSrxMnO3-δ is one of the most important perovskite oxides and has been extensively used as ORR electrocatalysts at room temperature [[33], [34], [35], [36]]. Furthermore, the B-site cation doping of La1-xSrxMnO3-δ was reported to improve the ORR activity at room temperature [[37], [38], [39], [40]]. For example, La0.8Sr0.2Mn1-xNixO3-δ (x = 0.2 and 0.4) displayed much higher ORR activity at room temperature than that of the pristine La0.8Sr0.2MnO3-δ (LSM) due to the formation of oxygen vacancies introduced by Ni doping [37]. Very recently, B-site Co-doped (La0.8Sr0.2)1-xMn1-xCoxO3-δ (x = 0.05 and 0.1) showed much higher ORR activity than LSM and A-site deficient LSM at room temperature in an alkaline medium [38].
In addition to the metal element doping, researchers also focus on the non-metal element doping in perovskite oxides to enhance the electrocatalytic performance at room temperature [[41], [42], [43], [44]]. For instance, P-doped SrCo0.95P0.05O3-δ (SCP) perovskite was developed as a high-performance electrocatalyst for oxygen evolution reaction (OER) in an alkaline solution due to the greatly improved electrical conductivity and stabilized perovskite structure [42]. Very recently, P-doped LSM perovskite oxides have been developed as ORR electrocatalysts at room temperature in an alkaline medium by using P as a non-metal dopant for the cation site (B site) [44]. However, to the best of our knowledge, the co-doping of one metal element and one non-metal element into a perovskite oxide as an ORR electrocatalyst at room temperature in an alkaline medium has not been reported.
Herein, we develop a co-doping strategy for improving the ORR activity and stability of LSM in an alkaline medium by simply using Sc3+ and P5+ as the functional dopants. The co-doped La0.8Sr0.2Mn0.95Sc0.025P0.025O3-δ (LSMSP) displays higher ORR activity and stability than LSM and single-element-doped LSM (La0.8Sr0.2Mn0.95Sc0.05O3-δ and La0.8Sr0.2Mn0.95P0.05O3-δ, which were denoted as LSMS and LSMP, respectively) due to the increased amounts of Mn3+ species and surface adsorbed oxygen species, an enlarged specific surface area, a reduced particle size, and the synergistic effect originated from the co-doping. Furthermore, this co-doped perovskite electrocatalyst showed a comparable activity and a better stability for ORR as compared with Pt electrocatalyst. This work can provide some useful guidelines for the design of high-performance electrocatalysts for ORR, which may be applicable in PEMFCs and metal-air batteries.
LSM, LSMP, LSMS and LSMSP samples were prepared by a sol-gel route [44]. Taking the LSMSP (0.02 mol) as an example, stoichiometric amounts of La(NO3)3⋅6H2O (6.9282 g), Sr(NO3)2 (0.8465 g), Mn(CH3COO)2⋅4H2O (4.6567 g), Sc2O3 (0.0346 g Sc2O3 was first dissolved by 0.5 M HNO3, 50 mL) and NH4H2PO4 (0.0660 g) were mixed in 500 mL deionized water. All reagents were purchased from Sinopharm Chemical Reagent Co., Ltd., analytical grade (99.9%). Following the addition of ethylenediaminetetraacetic acid (EDTA, 11.6896 g) and citric acid (CA, 18.8112 g), an aqueous ammonia solution (40 mL) was added to achieve a pH value of 6. The ratio of total metal ions/EDTA/CA was 1:1:2. After heating at 90 ℃ under stirring, a transparent gel was obtained and then heated at 250 ℃ for 5 h in air to form a solid precursor, which was further calcined in air at 800 ℃ for 5 h. Then the obtained powders were ground by agate mortar. The crystal structures of perovskite oxides were characterized by powder X-ray diffraction (XRD, D8 Advance Bruker) at room temperature. The microstructures and particle sizes were obtained by using a field-emission scanning electron microscope (FE-SEM, Hitachi-S4800). The elemental concentrations of the various perovskite oxides were investigated by energy-dispersive X-ray spectroscopy (EDX, S3400). Brunauer-Emmett-Teller (BET) specific surface areas were characterized by nitrogen adsorption-desorption tests (BELSORP II). The surface metal valence states and oxygen-related species were analyzed by X-ray photoelectron spectroscopy (XPS, PHI 5000 VersaProbe spectrometer).
The ORR performance was investigated by polarization curves and electrochemical impedance spectroscopy (EIS) on a rotating disk electrode (RDE, ALS Co., Ltd.) in a 3-electrode configuration by an electrochemical workstation (CHI760E) [44]. All ORR measurements were conducted by using a glassy carbon (GC, 4 mm diameter) supported electrocatalyst as the working electrode, a Pt counter electrode and a Ag/AgCl reference electrode (3.5 M KCl) in O2-saturated 0.1 M KOH. The electrolyte was bubbled with oxygen gas continuously during each ORR measurement. The Ag/AgCl (3.5 M KCl) potentials were converted to reversible hydrogen electrode (RHE) potentials by using the equation E(RHE) = E(Ag/AgCl, 3.5 M KCl) + 0.950 V based on the calibration method (Fig. S1 in Supporting Information). The electrocatalyst ink was prepared by sonicating a mixture of catalyst (10 mg), conductive carbon (10 mg), Nafion solution (5 wt%, 0.05 mL) and ethanol (0.5 mL) for about 30 min. The electrocatalyst ink (0.01 mL) was loaded on the top of the GC disk. Additional conductive carbon was not needed for the preparation of Pt/C ink. The perovskite oxide and 20 wt% Pt/C (purchased from Johnson-Matthey) loadings were 0.724 mg cm-2 (0.362 mgoxide cm-2) and 0.181 mg cm-2 (0.0362 mgPt cm-2), respectively. The polarization curves were recorded by linear sweep voltammetry (LSV) at a given rotation rate and a scan rate of 5 mV s-1 between 1.1 and 0.35 V vs. RHE in O2-saturated 0.1 M KOH. The durability tests were conducted by continuous potential cycling between 1.1 to 0.35 V vs. RHE at 5 mV s-1 and 1600 rpm in O2-saturated 0.1 M KOH. After 500 cycles, LSV was then conducted to obtain the long-term stability. The chronoamperometric tests were tested at 0.35 V vs. RHE and 1600 rpm in O2-saturated 0.1 M KOH.
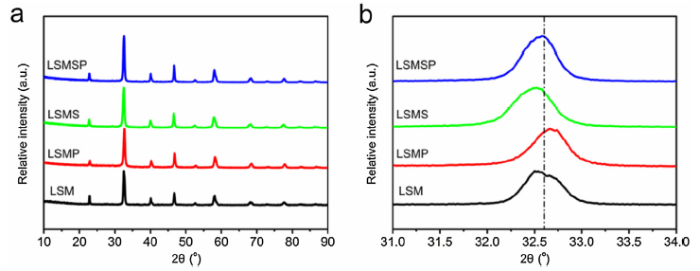
The phase structures of LSM, LSMP, LSMS and LSMSP were investigated by XRD as depicted in Fig. 1. All the diffraction patterns of the B site doped LSM perovskite oxides can be well indexed to be a cubic phase with a space group of Pm-3 m, which is the same as LSM, implying no phase change after the Sc and/or P doping. For LSMS, the XRD peaks shift to a lower angle, suggesting a lattice expansion due to the presence of the larger ionic radius of Sc3+ than those of Mn3+/Mn4+. In contrast, due to the smaller ionic radius of P5+, the diffraction peaks of LSMP shift slightly to a higher angle as compared with those of the pristine LSM. It is interesting that there are no obvious shifts in the diffraction peaks of LSMSP due to the balance between the lattice expansion and shrinkage caused by Sc3+ and P5+ doping, respectively. We also investigated the surface microstructures of LSM, LSMP, LSMS and LSMSP by SEM as shown in Fig. 2. As can be seen, LSMP, LSMS and co-doped LSMSP show much smaller particle sizes than that of LSM, implying more catalytic active sites for the ORR. As shown in Fig. S2, the BET specific surface areas of LSM, LSMS, LSMP and LSMSP samples were measured to be 6.92, 13.2, 27.4 and 29.8 m2 g-1, respectively. We propose that the increased specific surface areas of LSMS, LSMP and LSMSP samples are attributed to the reduced particle size.
Fig. 1. (a) XRD patterns and (b) magnified region at 2θ = 31°‒34° of La0.8Sr0.2MnO3-δ, La0.8Sr0.2Mn0.95P0.05O3-δ, La0.8Sr0.2Mn0.95Sc0.05O3-δ and La0.8Sr0.2Mn0.95Sc0.025P0.025O3-δ samples.
Fig. 2. SEM images of (a) La0.8Sr0.2MnO3-δ, (b) La0.8Sr0.2Mn0.95P0.05O3-δ, (c) La0.8Sr0.2Mn0.95Sc0.05O3-δ, and (d) La0.8Sr0.2Mn0.95Sc0.025P0.025O3-δ samples.
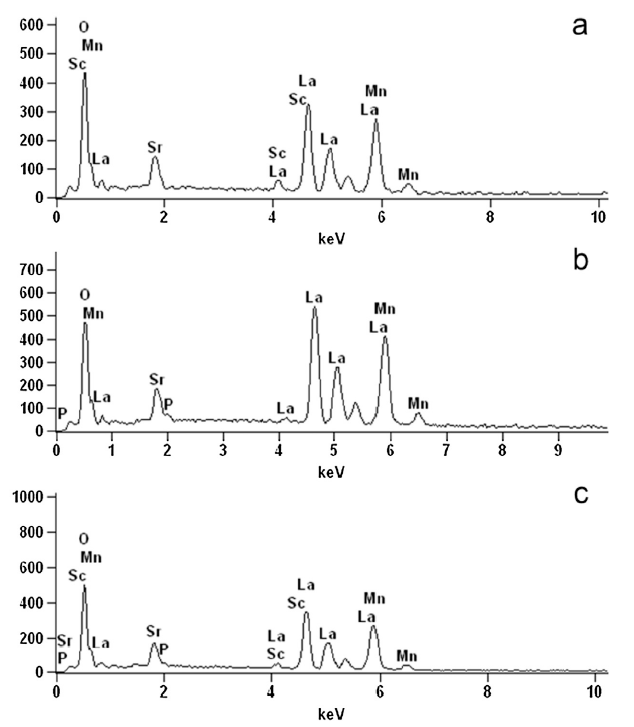
The Sc3+ and/or P5+ doping concentrations of the LSMP, LSMS and co-doped LSMSP were studied by EDX with results shown in Fig. 3. As can be seen, the Sc3+ and P5+ doping amounts in LSMS and LSMP were 0.0256 and 0.0249, respectively, which were very close to the theoretical value in the chemical formula (0.0250). In addition, the Sc3+ and P5+ doping amounts in LSMSP were 0.0128 and 0.0124, respectively, which were similar to the theoretical value (0.0125). The above results suggested that Sc3+ and/or P5+ were successfully doped into the LSM perovskite with well-controlled doping concentrations.
Fig. 3. EDX profiles of (a) La0.8Sr0.2Mn0.95Sc0.05O3-δ, (b) La0.8Sr0.2Mn0.95P0.05O3-δ, and (c) La0.8Sr0.2Mn0.95Sc0.025P0.025O3-δ samples.
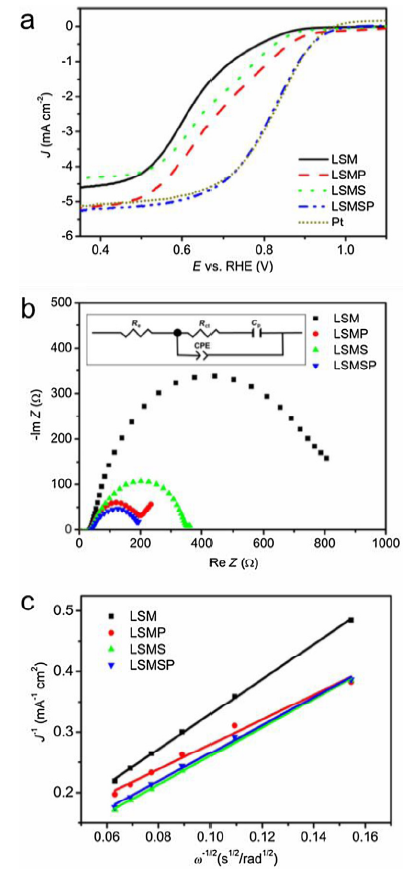
LSV tests at a rotation rate of 1600 rpm were used to investigate the ORR activity of various electrocatalysts as shown in Fig. 4(a). The LSMSP electrocatalyst displayed a superior ORR activity to LSM, LSMS and LSMP, which was comparable to Pt/C. More specifically, the onset potential (Eonset) and half wave potential (E1/2) values for LSMSP were 0.96 and 0.81 V vs. RHE, respectively, outperforming LSM (0.86 and 0.61 V vs. RHE), LSMP (0.95 and 0.67 V vs. RHE) and LSMS (0.89 and 0.65 V vs. RHE), which was comparable to the values for Pt (0.95 and 0.82 V vs. RHE). To further understand the ORR kinetics of the perovskite oxides, EIS measurements were implemented at 0.27 V as shown in Fig. 4(b). Based on the equivalent circuit composed of electrolyte resistance (Re), charge transfer resistance (Rct), a constant-phase element (CPE) and a pseudocapacitive element (Cp), the Rct values of LSM, LSMP, LSMS and LSMSP were 932, 155, 317 and 139 Ω, respectively, showing enhanced charge transfer capability of P-doped LSMSP and LSMP. To be specific, the charge transfer capability of these four samples follows the order of LSMSP > LSMP > LSMS > LSM, agreeing well with the ORR performance tendency shown in Fig. 4(a). We also investigated the electron transfer pathway of various electrocatalysts using the Koutechy-Levich (K-L) equation, as depicted in Fig. 4(c) [44]. The LSV curves obtained by various electrocatalysts at different rotation rates were shown in Fig. S3. The n values were 3.55, 3.84, 3.68 and 3.71 for LSM, LSMP, LSMS and LSMSP, respectively, suggesting that the doped LSM perovskites are more likely to undergo a 4-electron pathway to reduce oxygen to OH- in the ORR.
Fig. 4. (a) Linear sweep voltammetry curves of La0.8Sr0.2MnO3-δ, La0.8Sr0.2Mn0.95P0.05O3-δ, La0.8Sr0.2Mn0.95Sc0.05O3-δ, La0.8Sr0.2Mn0.95Sc0.025P0.025O3-δ and Pt/C at 1600 rpm in O2-saturated 0.1 M KOH, (b) EIS spectra of perovskite samples obtained at a frequency range of 100 kHz to 0.1 Hz at 0.27 V vs. RHE under a 10 mV AC potential bias in O2-saturated 0.1 M KOH, (c) The n values of various perovskite electrocatalysts based on the K-L equation at 0.35 V vs. RHE.
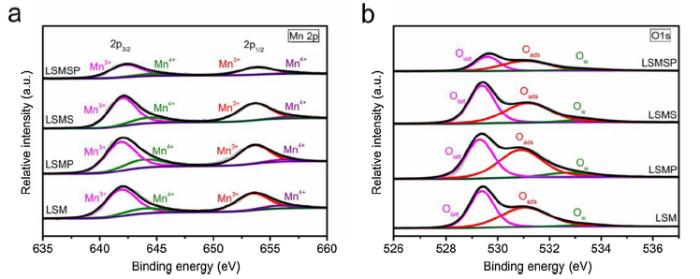
XPS was employed to study the valence states of the Mn ions and the oxygen-related species on the surfaces of LSM, LSMP, LSMS and LSMSP electrocatalysts. Based on the fitted Mn 2p XPS spectra as shown in Fig. 5(a), the peaks centered at ~640.9 and 652.6 eV matched with the Mn3+ species while the peaks at 642.8 and 654.3 eV corresponded to the Mn4+ species. As shown in Fig. 5(a) and Table S1, the average oxidation states of the Mn ions in LSM, LSMP, LSMS and LSMSP were 3.18, 3.21, 3.15 and 3.11, respectively, suggesting that more Mn3+ and oxygen vacancy was formed after Sc3+ doping. It was reported that the Mn3+ species was beneficial to achieving higher ORR activity than Mn2+ and Mn4+ [45]. The mixed valence states of Mn (Mn3+ and Mn4+) in these four electrocatalysts played an important role in promoting the charge transfer process to achieve high ORR activity [44,45]. The larger amount of Mn3+ in the co-doped LSMSP electrocatalyst contributed to its higher ORR activity than those of LSM, LSMP and LSMS. As shown in Fig. 5(b), the O 1s XPS spectra were fitted into three peaks of the surface oxygen-related species, including lattice oxygen (Olatt, ~529.3 eV), adsorbed oxygen (Oads, ~531.1 eV) and adsorbed water (Ow, ~533.1 eV) by the CasaXPS software (Version 2.3.18). The relative concentration of each oxygen species over the total amount of surface oxygen was estimated from the relative area of these fitted sub-peaks (detailed in Table S1). Obviously, LSMSP displayed the highest amount of Oads, which benefits the oxygen adsorption process in ORR. The above results highlight that the optimized Mn valence state, higher Oads amount, larger surface area and smaller particle size are main contributing factors for superior ORR activity of LSMSP electrocatalyst.
Fig. 5. (a) Mn 2p and (b) O 1s XPS spectra of La0.8Sr0.2MnO3-δ, La0.8Sr0.2Mn0.95P0.05O3-δ, La0.8Sr0.2Mn0.95Sc0.05O3-δ and La0.8Sr0.2Mn0.95Sc0.025P0.025O3-δ samples.
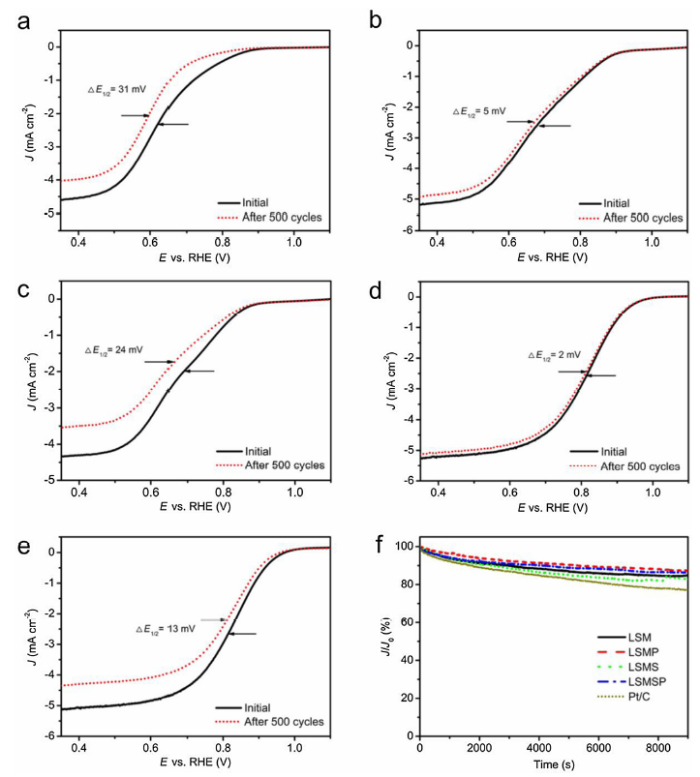
The durability of the ORR electrocatalysts is also critical for practical applications, which was investigated by testing the LSV curves before and after 500 potential cycles. As shown in Fig. 6, the E1/2 shifted negatively by only 2 mV, implying the superior durability of LSMSP electrocatalyst in alkaline medium introduced by Sc and P co-doping. In contrast, the E1/2 shifted negatively by 31, 5, 24 and 13 mV for LSM, LSMP, LSMS and Pt/C electrocatalysts, respectively. As shown in Fig. 6(f), the chronoamperometric tests (I‒t) suggested that ~87% of the initial ORR activity of LSMSP and LSMP electrocatalysts was retained after a 9000 s test, while the performance of LSM, LSMS and Pt/C electrocatalysts decreased to ~84%, ~83% and ~77% as compared with the initial performance under the same conditions, respectively.
Fig. 6. Linear sweep voltammetry curves of (a) La0.8Sr0.2MnO3-δ, (b) La0.8Sr0.2Mn0.95P0.05O3-δ, (c) La0.8Sr0.2Mn0.95Sc0.05O3-δ, (d) La0.8Sr0.2Mn0.95Sc0.025P0.025O3-δ and (e) Pt/C obtained before and after 500 potential cycles, (f) I‒t curves of various electrocatalysts in O2-saturated 0.1 M KOH.
In summary, we developed a Sc and P co-doped perovskite oxide as a new electrocatalyst with remarkable performance for oxygen reduction reaction in an alkaline electrolyte. The oxygen reduction activity of La0.8Sr0.2Mn0.95Sc0.025P0.025O3-δ electrocatalyst compared favourably to that of Pt/C, which was much higher than those of La0.8Sr0.2MnO3-δ, Sc doped La0.8Sr0.2MnO3-δ and P doped La0.8Sr0.2MnO3-δ electrocatalysts. More specifically, the La0.8Sr0.2Mn0.95Sc0.025P0.025O3-δ electrocatalyst showed a high oxygen reduction activity in terms of overpotential (Eonset of 0.96 V vs. RHE and E1/2 of 0.81 V vs. RHE) when compared with La0.8Sr0.2MnO3-δ (0.86 and 0.61 V vs. RHE), La0.8Sr0.2Mn0.95Sc0.05O3-δ (0.89 and 0.65 V vs. RHE) and La0.8Sr0.2Mn0.95P0.05O3-δ (0.95 and 0.67 V vs. RHE). Furthermore, La0.8Sr0.2Mn0.95Sc0.025P0.025O3-δ showed an improved stability for oxygen reduction reaction in the I‒t and cycling tests (9000 s and 500 cycles, respectively) compared to Pt/C, La0.8Sr0.2MnO3-δ, La0.8Sr0.2Mn0.95Sc0.05O3-δ and La0.8Sr0.2Mn0.95P0.05O3-δ. This remarkable oxygen reduction performance of La0.8Sr0.2Mn0.95Sc0.025P0.025O3-δ was attributed to the increased amounts of Mn3+ ions and Oads, enlarged surface area, reduced particle size and the synergetic co-doping effect, suggesting that the co-doping of P and Sc is an effective strategy in enhancing the electrocatalytic activity of La0.8Sr0.2MnO3-δ, which opens up new possibilities to develop other perovskite oxides for various electrocatalysis-based applications.
This work was financially supported by the National Natural Science Foundation of China (Nos. 21576135 and 21706129), the Youth Fund of Jiangsu Province (No. BK20150945), Program for Jiangsu Specially-Appointed Professors and the Funding from State Key Laboratory of Materials-Oriented Chemical Engineering (No. ZK201808). The authors also acknowledge the financial support of the Australian Research Council.
Supplementary material related to this article can be found, in he online version, at doi:https://doi.org/10.1016/j.jmst.2019.09.007.

 WeChat
WeChat
/
| 〈 |
|
〉 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}