



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Corrosion Behavior of X52 Anti-H2S Pipeline Steel Exposed to High H2S Concentration Solutions at 90 °C
[Liu Meng, Wang Jianqiu*  , Ke Wei, Han En-Hou]
, Ke Wei, Han En-Hou]
, Ke Wei, Han En-Hou]
|
|


Initial corrosion kinetics of X52 anti-H2S pipeline steel exposed to 90 °C/1.61 MPa H2S solutions was investigated through high temperature and high pressure immersion tests. Corrosion rates were obtained based on weight loss calculation. The corrosion products were analyzed by scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray diffraction (XRD) and electron probe micro-analysis (EPMA). The initial corrosion kinetics was found to obey the exponential law. With increasing immersion time, the main corrosion products changed from iron-rich mackinawite to sulfur-rich pyrrhotite. The corrosion films had two layers: an inner fine-grained layer rich in iron and an outer columnar-grained layer rich in sulfur. The corrosion film formed through the combination of outward diffusion of Fe2+ ions and inward diffusion of HS- ions. The variation of the corrosion products and compaction of the corrosion layer resulted in a decrease in the diffusion coefficient with increasing immersion time. The double-layered corrosion film formed after long time immersion acted as an effective barrier against diffusion.
Internal H2S corrosion of carbon steel is a major concern in oil and gas industry [1], [2] and [3]. Its understanding, prediction and control are key challenges to sound facilities design, operation and subsequent integrity assurance. The corrosion failures can lead to costly repairs, plant shutdowns, as well as health and environmental hazards. Along with deep exploitation of oil and gas, the H2S corrosion problem becomes much severer as the water salinity, the service temperature and the H2S partial pressure increase [2]. Almost 1/4 of Chinese natural gas reservoirs contain more than 1% H2S. Highly sour gas fields (H2S concentrations more than 5%) are mainly distributed in the Xuanhan–Dazhou area of Northeastern Sichuan [4]. The content of hydrogen sulfide in some wells is more than 10%, where the H2S partial pressure reaches about 3 MPa and the temperature reaches 95–115 °C in Puguang gas field [5]. The corrosion behavior and mechanism of mild steel are imperative for corrosion engineers to carry out the corrosion prevention design. However, only few investigations have been carried out under these high temperature and high H2S partial pressure conditions. Omar et al. [6] investigated the corrosion of carbon steel under simulated Kashagan field conditions. It was concluded that the average corrosion rates were relatively low in the range of 0.5–1.5 mm/year, and did not vary much with temperature or flow velocity, but there was a risk of localized corrosion attacks due to film breakdown. Through high temperature and high pressure corrosion simulation experiments, Zhang et al. [5] concluded that under 1–2 MPa H2S, the corrosion rate increased with elevating temperature up to 60 °C and then decreased gradually. Serious pitting corrosion was found at 30 °C. Temperature and partial pressure directly affected the morphology and composition of corrosion products, which in turn caused the change of corrosion rate and occurrence of localized corrosion. Kinetics of iron carbonate and iron sulfide scale formation in CO2/H2S corrosion was systematically investigated by Sun [7] using weight change method, and a mechanistic model for H2S corrosion was developed to predict the CO2/H2S corrosion rate, but this model did not include iron sulfide precipitation effects which always occurred under high temperature conditions, and it deviated a lot from the experimental data when extended to conditions with H2S partial pressure more than 10 bar [8]. As a result this model is unsuitable under high temperature and high H2S partial pressure conditions. Corrosion at higher H2S concentrations and moderate temperatures was reviewed by Smith et al. [2]. It was mentioned that there was only a limited volume of work available in literature that described the corrosion mechanism and the corrosion reactions that resulted in the formation of pyrrhotite in aqueous corrosion systems, especially at temperatures below 100 °C. Surface scale formation is one of the important factors governing the corrosion rate [9], and it is still unclear how does the variation in film composition affect the corrosion rate. The uncertain mechanism of H2S corrosion makes it difficult to model the iron sulfide layer formation and further to predict the corrosion rate of mild steel under these conditions. In this paper the initial corrosion behavior of commercial X52 anti-H2S pipeline steel was investigated in pure water under H2S partial pressure of 1.61 MPa at 90 °C. The evolution of weight loss with time was obtained and fitted. Morphology of corrosion products was obtained through scanning electron microscopy (SEM) observation. X-ray diffraction (XRD) was also used to identify the variation of the corrosion products during the corrosion process. The element distribution of the corrosion product was analyzed by electron probe micro-analysis (EPMA). The corrosion mechanism was also discussed according to the mechanism proposed by former literature.
The specimens were cut from commercial X52 anti-H2S pipeline steel with 108 mm outer diameter and 10 mm thickness. The chemical composition of this material is presented in Table 1 which meets the requirements of the API 5L standard. The microstructure and inclusions of test material as shown in Fig. 1 were investigated by SEM and energy dispersive X-ray analysis (EDX) after being polished and etched in a 2% Nital (nitric acid–ethanol) solution. As can be seen from Fig. 1(a), the microstructure consists of ferrite and pearlite with randomly distributed elongated manganese sulfides inclusions and small globular oxide inclusions, and this distribution and morphology of the non-metallic inclusions which are resisted for hydrogen induced cracking resistance are designed to meet the anti-H2S requirements. The microstructure at low magnification shows banded structure with ferrite and pearlite elongated along the rolling direction as shown in Fig. 1(b).
| Table 1. Chemical composition of X52 anti-H2S pipeline steel (wt%) |
 | Fig. 1. Microstructure of X52 anti-H2S pipeline steel: (a) inclusions, (b) banded structure of ferrite and pearlite. |
The immersion test was carried out in a 2.2 L autoclave made by Cortest Company. This autoclave was made using a Hastelloy C276 (UNS N10276) with designed temperature of 300 °C and designed pressure of 35 MPa. The testing solution was pure water dissolved with H2S.
All the test specimens of 30 mm × 20 mm × 3 mm in size, were cut from the material and wet-ground with silicon carbide papers up to 1000 grit and then ultrasonically cleaned with alcohol for 5 min. After being weighed, they were immersed in the test solution. After being deoxygenated by bubbling N2 for 2 h, the test autoclave was heated to test temperature 90 °C, and then charged with H2S to the target pressure of 1.61 MPa. The immersion test lasted for 2, 6, 18, 24, 56, 96 and 232 h, respectively.
After immersion tests, samples were weighed again after being washed in 0.2% ethylene diamine tetraacetic acid (EDTA) solution and dried. The evolution of the weight loss with time was calculated through . The morphology of corrosion products was observed by SEM. The crystal structure of the corrosion products was analyzed by XRD. The element distribution of the corrosion films was also analyzed by EPMA after being mounted in epoxy.
where Ris the corrosion rate, mm/year; M1is the weight of the test samples before immersion test, g; M2is the weight of the test samples after the corrosion products was removed, g; Sis the area of the test samples, cm2; Tis the immersion time, h; Dis the density of X52 anti-H2S pipeline steel, 7.86 × 103kg/m3.
A gold layer of 5 mm × 10 mm × 20 nm in size was sputtered on several samples with dimensions of 10 mm × 15 mm × 3 mm using a Gatan 682 model precision etch coating system, and the sputtering area was in the middle of the sample and the rest area of the sample was sealed by silver paper. These samples were tested together with other samples to investigate the corrosion film formation process in the 96 h immersion test. In order to confirm the presence of different iron sulfides, the corrosion film formed after immersion for 2 h was scraped from the sample surface, and then the products were dispersed in ethanol using ultrasonic. The corrosion products were collected using a copper net and then analyzed by TEM (FEI Technai G2 F30).
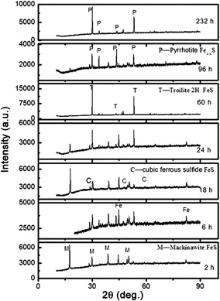
Fig. 2 presents the phase variation of the corrosion products at different immersion time through XRD analysis. As seen in Fig. 2, there are mainly 4 types of corrosion products: mackinawite (FeS1- x), troilite (FeS), cubic ferrous sulfide (FeS) and pyrrhotite (Fe1- xS). The Pourbaix diagrams [10], [11], [12], [13] and [14] developed for prediction of iron sulfide formation under different conditions illustrate that troilite and pyrrhotite are thermodynamically stable products under the test conditions. Mackinawite is a thermodynamically semi-stable form of FeS that forms on the steel surface under conditions where iron sulfide is still soluble in the bulk solution [7], and it is acknowledged that mackinawite initially is formed by a fast solid-state reaction. The other iron sulfide such as cubic ferrous sulfide, troilite and pyrrhotite are formed mainly through precipitation [15]. And the mackinawite formation kinetics is much faster than those of other iron sulfides [16]. So during the first 24 h immersion, the corrosion products were constructed mainly by mackinawite with minor troilite and cubic ferrous sulfide, and mackinawite existed during the whole immersion time. The solubility for mackinawite is much higher than that of troilite and pyrrhotite [17].
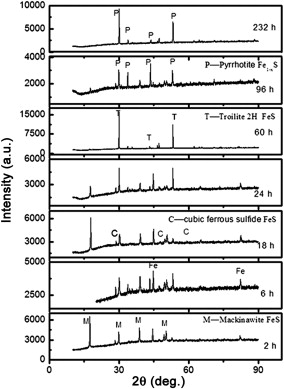 | Fig. 2. XRD patterns for the corrosion products formed after different immersion time at 90 °C, in 1.61 MPa H2S solution. |
As immersion time increased, mackinawite was formed and dissolved, and the nucleation and growth of troilite and pyrrhotite was gradually dominated. As a result, the major corrosion product varied from iron-rich mackinawite to sulfur-rich troilite and pyrrhotite gradually. Cubic ferrous sulfide as a metastable iron sulfide only found in corrosion products, can easily transform to mackinawite. Once troilite forms, it grows at the expense of cubic ferrous sulfide [15], so cubic ferrous sulfide was not detectable after 232 h immersion.
Fig. 3(a–c) shows the TEM images with electron diffraction patterns of the iron sulfides formed after 2 h immersion. As can be seen from Fig. 3, amorphous FeS also forms except for mackinawite and troilite. It is acknowledged that amorphous FeS first forms in the corrosion process and it can easily transform to mackinawite [10] and [16].
 | Fig. 3. TEM micrographs with electron diffraction patterns of the iron sulfide formed on X52 anti-H2S pipeline steel after 2 h immersion at 90 °C, in 1.61 MPa H2S solution: (a) mackinawite, (b) troilite, (c) amorphous FeS. |
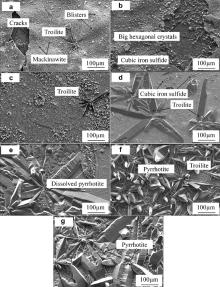
Fig. 4(a–g) shows the SEM images of the iron sulfides formed after different immersion time. After 2 h immersion, a layer of corrosion products, small tetragonal mackinawite, has already uniformly covered the sample surface, and several big hexagonal troilite crystals form on top of the corrosion scale. As the corrosion scale mainly constructed by mackinawite grew thicker, it cracked and peeled off due to the stress as a result of volume effect [15]. Therefore the corrosion scale is very loose, and has many blisters and even cracks as shown in Fig. 4(a). Once the corrosion scale cracked and fresh surface was exposed, the corrosion process was accelerated. More Fe2+ was released into solution, which would favor the formation of iron sulfide through precipitation. So beneath the cracks big hexagonal crystals are formed as shown in Fig. 4(b). After 6 h immersion, the corrosion scale cracks more severe and more hexagonal crystals form underneath the cracks. As the immersion time reaches 18 h, an integrated new formed corrosion scale forms after the former scale cracks and peels off, and more big hexagonal products form on top of this newly formed scale. The corrosion products change to mainly big hexagonal crystals as the reaction time increases, which is in agreement with the XRD results. Also the corrosion products become much denser as more thermodynamically troilite and pyrrhotite precipitate, although the big hexagonal products begin to dissolve as seen in Fig. 4(e).
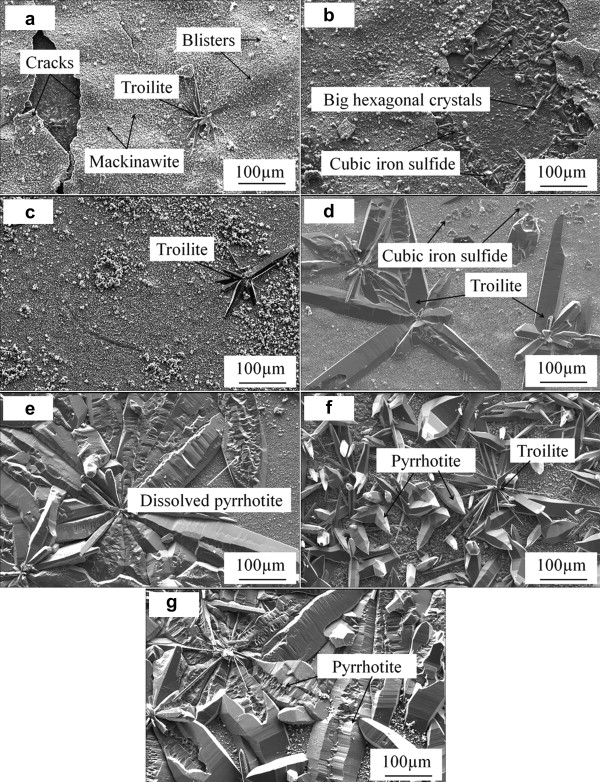 | Fig. 4. SEM images of corrosion products of X52 anti-H2S pipeline steel after immersed at 90 °C, in 1.61 MPa H2S solution: (a) 2 h, (b) 6 h, (c) 18 h, (d) 24 h, (e) 60 h, (f) 96 h, (g) 232 h. |
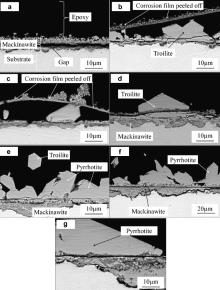
Fig. 5(a–g) shows the cross-section images of the corrosion film obtained by back scattered electron (BSE). It shows general corrosion without pitting or hydrogen induced cracking. The initially formed corrosion scale consists of mackinawite cracked and peeled off which is also seen in Fig. 5(b) and (c). The corrosion scales except for that in Fig. 5(a) consist of two layers: an inner fine-grained layer and an outer columnar-grained layer. Fig. 5(a) shows that there is a gap between the outer layer and the inner layer. This is because the adherence of the scale is too weak to keep the outer layer attached on the substrate and resist the stress introduced during mounting in epoxy [18]. Both layers become thicker and more big hexagonal products form with increasing immersion time.
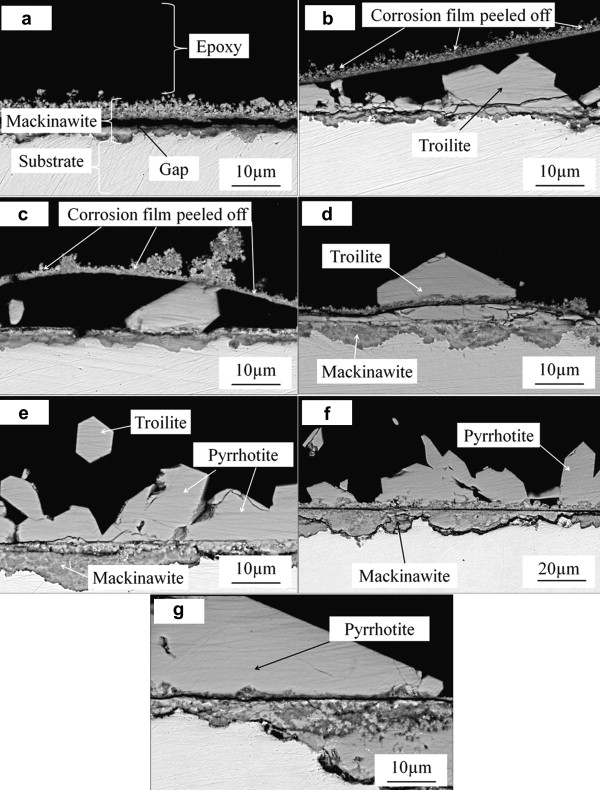 | Fig. 5. Cross-section images of the corrosion film of X52 anti-H2S pipeline steel after immersed at 90 °C, in 1.61 MPa H2S solution: (a) 2 h, (b) 6 h, (c) 18 h, (d) 24 h, (e) 60 h, (f) 96 h, (g) 232 h. |
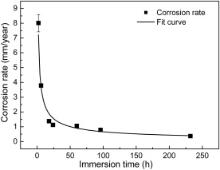
The corrosion rate at different immersion time is shown in Fig. 6. It is commonly acknowledged that the specific corrosion mechanism determines the corrosion rate of a material under a certain environment. The corrosion film acted as a diffusion barrier for the species involved in the corrosion process can slow down the corrosion process and prevent the underlying steel from further dissolution. As the corrosion film grows in density as well as in thickness, the corrosion rate will decrease if the corrosion products show perfect protective effect. The corrosion resistance of iron sulfides follows a sequence of mackinawite < troilite < pyrrhotite < pyrite [6], [15] and [18]. As shown in Fig. 2 and Fig. 4, during the first 24 h, the major corrosion product is mackinawite, and mackinawite shows little protection because it forms a loose and porous corrosion film. As a result the diffusion of electrochemical reaction species such as Fe2+ and HS– is easier through this kind of corrosion film. Some investigations [19] indicate that mackinawite can even accelerate corrosion rate. So the corrosion rate is very rapid during the first 18 h immersion. As more corrosion resistants such as troilite and pyrrhotite precipitate, the corrosion rate decreases dramatically from 8.02 to 1.12 mm/year after 24 h immersion. When the major products change to troilite and pyrrhotite after 60 h, the corrosion rate decreases slowly with increasing immersion time. The kinetics of corrosion was found to obey the exponential law which was always found in atmosphere corrosion. After curve fitting, exponential decay equation was obtained:
where Crepresents the corrosion rate, mm/year; krepresents the rate constant, mm/year; trepresents the immersion time, h; nis the time constant. The parameter kprovides a criterion for gauging short-term corrosion susceptibility. In our test, the kvalue is 11.02, indicating a rapid initial corrosion rate. The nvalue gives a measure of the resistance to transport process within the corrosion products once it forms [20]. The value for nis -0.61, meaning that the corrosion film acts as an effective barrier against diffusion, and this mainly results from a decrease in the diffusion coefficient with time through recrystallization and compaction of the corrosion layer [21]as seen in Fig. 2and Fig. 5.
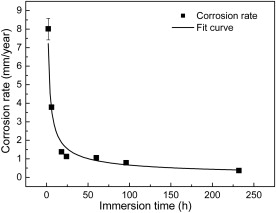 | Fig. 6. Corrosion rate of X52 anti-H2S pipeline steel as a function of the immersion time at 90 °C, in 1.61 MPa H2S solution. |
The diffusion of reaction species such as Fe2+ and HS- ions through corrosion product film is an important factor which affects the corrosion mechanism. It is assumed that Fe2+ ions diffuse through the corrosion film from the steel/corrosion film interface to the corrosion film/solution interface to react with HS- and H2S [18].
In the 96 h test, a gold marker initially sputtered on part of the metal surface was used to investigate the corrosion film formation process. As seen in Fig. 7, the gold marker representing the original sample surface is located at the interface between the inner and outer layers of the iron sulfide corrosion product. Thus the fine-grained inner layer must form by the inward migration of sulfide ions. The mass diffusion process includes the HS- and H2S species through the defects such as crevices and grain boundaries of the corrosion products towards the steel/corrosion film interface, since sulfide-ion migration in the lattice of iron sulfide is known to be very slow. This is in agreement with the experiment results obtained in H2S and H2 mixture [22]. The inward migration of sulfide ions leads to iron-rich sulfide mackinawite formation near the steel/corrosion film interface. The outward migration of Fe2+ ions leads to sulfur-rich troilite or pyrrhotite precipitation near the corrosion film/solution interface. It is known that ferrous ions in the pyrrhotite crystal diffuse more rapidly along the c-crystallographic direction [23], so the favorably oriented grains start growing along the preferred c-crystallographic direction as seen in Fig. 4(e–g), eventually establish the columnar-grained outer layer.
 | Fig. 7. Cross-section image of the corrosion film of X52 anti-H2S pipeline steel with an initially sputtered gold marker after 96 h immersion at 90 °C, in 1.61 MPa H2S solution. |
Fig. 8 shows the element distribution of Fe, S and O across the corrosion film formed after 96 h immersion through EPMA analysis. The element distribution shows that the inner layer is rich in iron and the outer layer is rich in sulfur, probably indicating that the inner layer consists of mackinawite and the outer layer consists of troilite and pyrrhotite. As seen in Fig. 8, a thin layer rich in oxygen exists in the corrosion frontier toward the steel substrate. According to the diffusion process mentioned above, the sulfide concentration gradually decreases towards the steel surface, and competitive formation of iron sulfide and iron oxide (probably magnetite, Fe3O4) takes place close to the steel surface where the sulfide concentration is low [24]. The existence of these oxides also indicates that the corrosion scale acts as the effective diffusion barrier and slows down the corrosion rate as proved in Eq. .
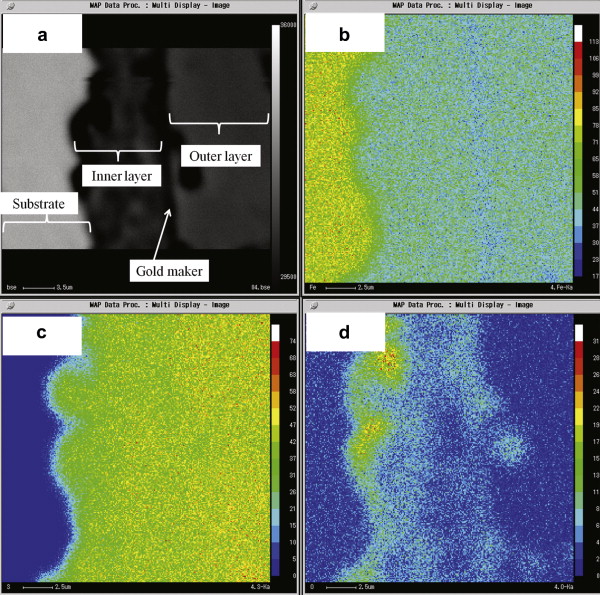 | Fig. 8. EPMA images of element distribution at the cross-section of the corrosion film on X52 anti-H2S pipeline steel with gold marker after 96 h immersion at 90 °C, in 1.61 MPa H2S solution: (a) cross-section of the film, (b) distribution of Fe, (c) distribution of S, (d) distribution of O. |
This work was supported by the National Natural Science Foundation of China (No. 51025104).
| 1. |
|
| 2. |
|
| 3. |
|
| 4. |
|
| 5. |
|
| 6. |
|
| 7. |
|
| 8. |
|
| 9. |
|
| 10. |
|
| 11. |
|
| 12. |
|
| 13. |
|
| 14. |
|
| 15. |
|
| 16. |
|
| 17. |
|
| 18. |
|
| 19. |
|
| 20. |
|
| 21. |
|
| 22. |
|
| 23. |
|
| 24. |
|