

1. Introduction
The energy dilemma and severe environmental pollution induced by the excessive consumption of traditional fuels have triggered the exploration for clean and renewable energy [1,2]. Hydrogen is an ideal substitute for fossil fuels because of its high calorific value and environmentally friendly combustion products. Hydrogen evolution reaction (HER) is deemed as a hopeful method for hydrogen preparation through electrocatalytic water splitting. Active and robust HER electrocatalysts are indispensable for triggering the water-splitting reaction. Although platinum currently exhibit the most active activity for electrocatalytic HER, but its large-scale development is limited by its low abundance and exorbitant price [3]. Therefore, the search for highly active and nonprecious catalysts for HER is crucial.
Recently, transition metal phosphides (TMPs) have received considerable attention due to their unique catalytic mechanism, which is similar to that of hydrogenase [4,5]. Phosphorus atoms present a strong electronegativity that attracts electrons from metal atoms and optimizes electronic structures [[6], [7], [8]]. The combination of transition metals and phosphorus will exhibit stronger metallic, which favors the improvement of catalyst conductivity. However, TMPs remain limited by their high overpotential because of the improper binding energy of the reaction intermediate and poor electron transfer kinetics [[9], [10], [11]]. In addition, as the extra activated water dissociation step requires the delicate design of catalytic materials, most TMPs possess lower activity and poorer stability in alkaline medium than in acidic medium [9,12,13].
Transition metal oxides (TMOs) are candidates for HER catalysts given their good stability over a wide range of electrochemical windows in bases [14]. We have previously reported the synergistic effect of the double-active metals Co and Mo, which exhibit good electrocatalytic activity [[15], [16], [17]]; on the other hand, Co-Mo bimetallic oxides are well tolerated in alkaline solutions [18,19]. We also observed the near-zero Gibbs free energy (ΔGH*) in the heterostructure interface of these oxides. This characteristic reflects high hydrogen adsorption efficiency.
Therefore, we designed a CoP/CoMoO4/carbon cloth (CC) heterostructure that combines CoP and CoMoO4 to fully exploit the outstanding catalytic activity of CoP and the excellent tolerance of CoMoO4 in alkaline media. It was obtained by reacting the precursor Co3O4@CoMoO4/CC with red phosphorus through the chemical vapor deposition (CVD) system. CoMoO4 not only increases the corrosion resistance of catalysts in alkaline media but also provides electron transport channels that will enhance catalyst conductivity. CoP/CoMoO4/CC exhibited an optimal overpotential of 81 mV at 10 mA cm-2 with a low Tafel slope of 69 mV dec-1. The negligible voltage change in the stability test indicates excellent durability in alkaline solution.
2. Experimental
2.1. Synthesis of Co3O4@CoMoO4/CC
All chemicals were of analytical reagent and used without further treated.
CC was cut into a 1 cm × 2 cm rectangle and sonicated in concentrated nitric acid for 10 min to improve hydrophilicity. Then, the carbon cloth was ultrasonicated for 15 min in acetone, deionized water, and ethanol and dried at 60 °C for 2 h. Next, 0.119 g CoCl2·6H2O and 0.177 g (NH4)6Mo7O24 were dissolved to 30 ml Mini-Q water under constant magnetic stirring. The solution was then transferred to a 50 ml Teflon reactor and incubated at 120 °C for 8 h in an electric furnace. The resulting purple substrate was carefully removed and washed with water and ethanol. Co3O4@CoMoO4/CC was obtained through annealing at 400 °C for 1 h in N2 atmosphere.
2.2. Synthesis of CoP@CoMoO4/CC
Red phosphorus (200 mg) was placed in the upstream of the tube furnace. Co3O4@CoMoO4/CC was placed downstream and heated at 500 °C for 1 h in Ar atmosphere. The CoP@CoMoO4/CC was obtained from the furnace after cooling to room temperature. For comparison, we increased the reaction temperature to 600 °C (CoMoP-600), 700 °C (CoMoP-700), and 800 °C (CoMoP-800) without changing other conditions.
2.3. Synthesis of CoP/CC
A total of 0.582 g Co(NO3)2·6H2O and 0.6 g urea were added to 20 ml deionized water to form a pink solution. After constant magnetic stirring, the pretreated CC and the solution were decanted into a 50 ml Teflon-lined reactor and heated at 120 ℃ for 6 h in an autoclave. After the reaction, the Co(OH)x/CC precursor was rinsed with deionized water and ethanol for several times and dried at 60 ℃ for 8 h. The conditions for subsequent phosphating were the same as those for CoP@CoMoO4/CC.
2.4. Synthesis of CoMoO4/CC
To prepare CoMoO4/CC, 0.091 g Co(NO3)2·6H2O and 0.605 g Na2MoO4·H2O were dispersed into 25 ml deionized water. Then, the resultant mixture was transferred to a Teflon-lined stainless autoclave at 140 ℃ for 4 h. After the reaction, the CC with purple precipitates was removed, cleaned with water and ethanol, and then dried in an autoclave at 60 ℃ for 8 h. The as-prepared CC was placed in a quartz tube furnace at 400 ℃ for 1 h with N2 flow to obtain CoMoO4/CC.
2.5. Material characterization
Crystal structure data were characterized with Bruker D8 Advance X-ray diffractometer (XRD) with Cu-Kα radiation (λ =1.54 Å). Samples morphology was studied by using Hitachi S-4800 scanning electron microscope (SEM), FEI Titan G260-300 transmission electron microscopy (TEM). The X-ray photoelectron spectra (XPS) were acquired by using AXIS SUPRA-Kratos with Al excitation resource. Raman measurements were carried out on Renishaw InVia. The Brunner-Emmett-Teller (BET) were recorded on QuadraSorb station 1.
2.6. Electrochemical measurements
The HER performance of all samples was determined on a electrochemical workstation (Bio-Logic VMP-300) in a standard three-compartment cell by using a standard Hg/HgCl2 electrode as reference electrodes and a graphite rod as counter electrodes, respectively.
Linear sweep voltammetry (LSV) was recorded with a scan rate of 2 mV s-1, electrochemical impedance spectroscopy (EIS) was carried out under the frequency range of 100 kHz to 100 MHz and an applied voltage of 10 mV. All electrochemical measurements were performed in 1 M KOH solution. All the LSV data were obtained directly and without IR-corrected.
3. Results and discussion
CoP@CoMoO4/CC was obtained through the hydrothermal method and subsequent phosphating. Fig. 1 illustrates the preparation process. First, Co3O4@CoMoO4/CC was grown on the CC through hydrothermal method. The product was dehydrated though calcination in a tube furnace. The tube furnace was also used for subsequent phosphating reaction. In brief, 200 mg red phosphorus was deposited in a quartz boat at the upstream side. The as-synthesized Co3O4@CoMoO4/CC was placed at the downstream side of the furnace and allowed to anneal at 500 °C for 1 h under Ar atmosphere with a heating speed of 10 °C/min to obtain CoP@CoMoO4/CC.
Fig. 1.

Fig. 1.
Synthesis of Co3O4@CoMoO4/CC and CoP@CoMoO4/CC.
To distinguish the sample peaks, we scraped the prepared Co3O4@CoMoO4/CC and CoP@CoMoO4/CC from CC. Figs. S1 and 2 show the XRD patterns of Co3O4@CoMoO4/CC and CoP@CoMoO4/CC, respectively. After reacting with red phosphorus, the peak of Co3O4 was replaced by that of CoP (Fig. S1). The diffraction peaks at 31.6°, 32.2°, 36.8°, 48.2°, and 52.1° can be assigned to the (011), (002), (102), (211), and (103) lattice planes of CoP (JCPDF 29-0497), respectively. For both samples, the peaks at the 2θ values of 26.5°, 28.2°, 38.8°, and 53.7° belong to the diffraction from the (002), (-311), (-222), (400), and (-531) planes of CoMoO4 (JCPDF 21-0868), respectively. These results prove that only Co3O4 has reacted with red phosphorus to form CoP, and that CoMoO4 remained unchanged during phosphorization. To further understand the compositional changes in the reaction, we increased the reaction temperature of the precursor and red phosphorus to 600 °C, 700 °C, and 800 °C without changing other conditions. As shown in Fig. S2, the peak intensity of CoMoO4 gradually weakened with the increase in temperature. When the temperature reached 800 °C, the peak of CoMoO4 disappeared completely, and the appearance of a new MoP (JCPDF 24-0771) peak indicated that CoMoO4 has been decomposed into CoP and MoP at 800 ℃ for 1 h.
Fig. 2.
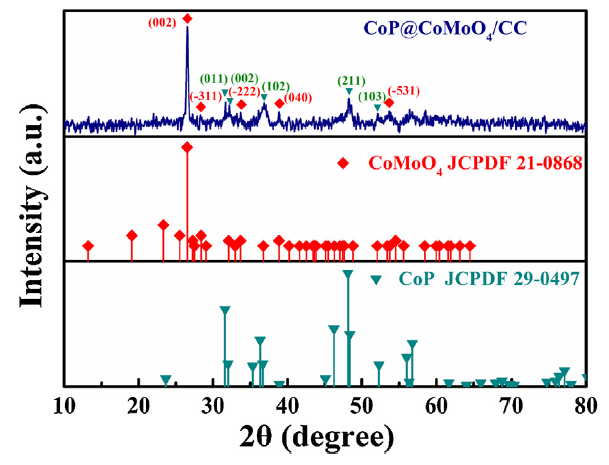
Fig. 2.
XRD pattern of CoP@CoMoO4/CC.
As shown in Fig. 3, the morphology of the products were conducted through scanning electron microscopy (SEM). The precursor Co3O4@CoMoO4/CC was a honeycomb structure assembled from nanosheets and nanowires structure. After reacting with red phosphorus at 500 ℃ for 1 h, CoP@CoMoO4/CC maintained the morphology of the precursor. However, the sharp nanowires became smooth because of their reaction with red phosphorus. Fig. S4 shows the SEM images of CoMoP-600, CoMoP-700, and CoMoP-800.
Fig. 3.

Fig. 3.
SEM images of Co3O4@CoMoO4/CC (a-c) and CoP@CoMoO4/CC (d-f).
The morphology of CoMoP-600 was almost unchanged compared with that of CoP@CoMoO4/CC. The boundary between the nanosheet and nanowire has become inconspicuous in CoMoP-700. After reacting at 800 ℃ for 1 h, the nanowires and nanosheets have become invisible and transformed into nanoparticles. Considering the previous XRD, this effect may be attributed to the reaction of the nanosheet with red phosphorus at high temperatures, making it unable to support the nanowires.
Co3O4@CoMoO4/CC and CoP@CoMoO4/CC were investigated by Raman spectroscopy to further study the composition information. As shown in Fig. S5, the strong intensity peaks at 1361 and 1594 cm-1 can be indexed to the D and G bands of the CC [20], respectively. The Raman modes centered at 340.1, 805.8, and 881.9 cm-1 in Figs. S2(a) and S2(b) correspond to the stretching vibration of Mo-O-Co in CoMoO4 [21]. In the Raman spectrum of Co3O4@CoMoO4/CC shown in Fig. S2(b), the peaks at 478.8, 522.3, 592.9 and 674.5 cm-1 were from the Eg, F2g, F2g, and A1g models of Co3O4 [22], respectively. All these peaks were absent from the Raman spectrum of CoP@CoMoO4/CC. These findings are in accordance to the conclusions drawn from the XRD patterns.
TEM was used to investigate the structural details of Co3O4@CoMoO4/CC and CoP@CoMoO4/CC. As shown in Fig. 4a-c, the nanowires and nanosheets were interdigitated in Co3O4@CoMoO4/CC. The high-resolution TEM (HRTEM) of nanowires showed that the lattice fringe of the nanowire reached 0.202 nm, which is consistent with the (400) plane of Co3O4. The HRTEM image obtained from the nanosheets showed that the lattice fringe totaled 0.230 nm, which is consistent with the (040) plane of CoMoO4. The HRTEM images in Fig. 4e and f illustrate that CoP@CoMoO4/CC featured interplanar distances of 0.231 and 0.245 nm, which correspond to the (040) plane of CoMoO4 and the (102) plane of CoP, respectively. These results indicate that Co3O4 in the precursor Co3O4@CoMoO4/CC was converted into CoP through the reaction with red phosphorus, and the lack of involvement of CoMoO4 in the reaction may be attributed to the better stability properties of the bimetallic substance than those of single-metal oxides.
Fig. 4.

Fig. 4.
TEM (a, b) and HRTEM images (c) of Co3O4@CoMoO4/CC; TEM (d) and HRTEM images (d, e) of CoP@CoMoO4/CC. STEM and elemental maps are shown in (g-k).
XPS was used to characterize the samples to gain additional information about the chemical composition and electronic state of Co3O4@CoMoO4/CC and CoP@CoMoO4/CC. Fig. 5b presents the high-resolution Co 2p spectrum. The Co 2p3/2 spectrum of Co3O4@CoMoO4/CC can be deconvoluted into Co2+ (781.2 eV) and Co3+ (779.6 eV) with a satellite peak (786.9 eV) [23,24]. In the Co 2p3/2 spectrum of CoP@CoMoO4/CC, the increase in the proportion of Co3+ (781.8 eV) peaks was accompanied by a decrease in the proportion of Co2+ (784.7 eV) peaks. These effects were due to the reaction of the precursor Co3O4 with phosphorus vapor and convert Co3O4 to CoP. The XPS spectrum of Mo 3d can be deconvoluted into four peaks was shown in Fig. 4c. two peaks at 231.7 and 234.8 eV in the Co3O4@CoMoO4/CC spectrum can be attributed to Mo6+. The presence of the peak of Mo6+ in CoP@CoMoO4/CC confirmed that CoMoO4, which contains Mo, failed to react with red phosphorus. In addition to Mo6+, Mo5+ peaks at 233.4 and 230.2 eV. Mo5+ may be attributed to the reduction of Mo6, as shown by previous reports on CoMoO4 [23,25]. The high-resolution P 1p spectra are shown in Fig. 5(d). The P 2p spectra of CoP@CoMoO4/CC can be deconvoluted into three peaks. The peak at 129.8 and 130.6 eV can be attributed to P 2p1/2 [12,26], which can be assigned to a metal-phosphorus bond such as CoP. The binding energy of 133.9 eV is generally associated with PO species, probably due to the oxidation of a small amount of P on the sample surface by oxygen in the air [27].
Fig. 5.

Fig. 5.
XPS survey spectra of Co3O4@CoMoO4/CC and CoP@CoMoO4/CC. (a) Co 2p, (b) Mo 3d, (c) O 1s, and (d) P 2p.
The electrochemical properties in 1 M KOH were also investigated to evaluate the HER performance of CoP@CoMoO4/CC. For comparison, bare CC, CoP/CC, CoMoO4/CC, Pt/C, CoP@CoMoO4/CC, and CoMoP-800 were measured by using the same method. It showed that the bare carbon cloth has negative HER activity in Fig. 5(a). CoP@CoMoO4/CC exhibited an outstanding HER activity. The overpotentials of CoP@CoMoO4/CC at 10 and 20 mA·cm-2 reached 81 and 121 mV, respectively, which are considerably lower than those of Co3O4@CoMoO4/CC (η10 = 255 mV, η20 = 296 mV), CoP/CC (η10 = 94.1 mV, η20 = 233 mV), CoMoO4/CC (η10 = 298 mV, η20 = 367 mV), and CoMoP-800 (η10 = 196 mV, η20 = 230 mV).
The Tafel slope is an important indicator for the evaluation of catalyst kinetics [28]. Commercial Pt/C has a minimum Tafel slope of 32 mV dec-1, which is indicative of first-rate reaction kinetics. The Tafel slope of CoP@CoMoO4/CC exhibited a value of 65 mV dec-1, which is optimal than that of the Tafel slopes of Co3O4@CoMoO4/CC (147 mV dec-1), CoP/CC (123 mV dec-1), CoMoO4/CC (167 mV dec-1), and CoMoP-800 (105 mV dec-1), indicating the fast kinetics for HER in alkaline solutions (Fig. 6[b]). The multipotential step technique was used to acquire the Tafel slope of CoP@CoMoO4/CC (Fig. S6). This finding agrees with the Tafel slope calculated from the LSV curve. Activity enhancement was mainly attributed to the enlargement of the active surface area and the increased electron transfer rate.
Fig. 6.

Fig. 6.
(a) LSV curves of Co3O4@CoMoO4/CC, CoP@CoMoO4/CC, CoP/CC, bare CC, CoMoO4/CC, commercial Pt/C, and CoMoP-800. (b) Corresponding Tafel plots. (c) Cdl measurements of Co3O4@CoMoO4/CC, CoP@CoMoO4/CC, CoP/CC, and CoMoO4/CC, and CoMoP-800. (d) Nyquist plots (at η = 250 mV). (e) Long-term stability testing of CoP@CoMoO4/CC at different current densities.
As shown in Fig. 6(c), the electrochemical double-layer capacitance (Cdl) at different scan rates of 10 mV s-1 to 60 mV s-1(Fig. S7) was measured to investigate the electrochemical active surface area (ECSA) [29], which is related to the number of exposed surface active sites. The Cdl of CoP@CoMoO4/CC (10.7 m F cm-2) is higher than 9.7 m F cm-2 for CoP/CC, 7.3 m F cm-2 for Co3O4@CoMoO4/CC, 0.9 m F cm-2 for CoMoO4/CC, and 3.1 m F cm-2 for CoMoP-800. The ECSA of samples was calculated through Cdl [30,31]. As shown in Table S1, the ECSA of CoP@CoMoO4/CC (178.3 cm2) is larger than CoP/CC (161.7 cm2), Co3O4@CoMoO4/CC (121.6 cm2), CoMoP-800/CC (51.7 cm2) and CoMoO4/CC (15.0 cm2). A large ECSA value indicates that numerous active sites are exposed. The specific surface area was analyzed by BET measurements (Fig. S10). The specific surface of CoP@CoMoO4 (12.22 m2 g-1) is larger than CoP (11.21 m2 g-1). With the same electrode area, the larger the specific surface area, the faster the gas escape [32]. Although the specific surface area of CoMoO4 (28.66 m2 g-1) and Co3O4@CoMoO4 (80.32 m2 g-1) is larger than CoP@CoMoO4, but in the heterostructure of CoP@CoMoO4, CoP has excellent catalytic activity and electrical conductivity, and CoMoO4 improves the specific surface area of the catalyst. Thus, CoP@CoMoO4/CC provided additional active sites that could improve the HER performance.
EIS was conducted to reveal the reaction kinetics of the electrode surface [33]. Charge transfer resistance (Rct) is related to the diameter of semicircles in the high-frequency zone. Small-diameter semicircles indicate the fast charge transfer of the electrode and enhanced reaction kinetics [34]. The smaller Rct (2.7 Ω) of CoP@CoMoO4/CC than those of CoP/CC (41.8 Ω), Co3O4@CoMoO4/CC (26.7 Ω), CoMoO4/CC (47.7 Ω), and CoMoP-800 (16.4 Ω) suggests the fast electron transport within CoP@CoMoO4/CC. The Rct of Co3O4@CoMoO4/CC is considerably smaller than that of CoP/CC and CoMoO4/CC. This result indicates that the nanowire-nanosheet crosslinked honeycomb structure could improve the charge transfer rate, thus enhancing the HER kinetics. CoP, which features better conductivity than Co3O4, benefitted the reduction of the Rct of CoP@CoMoO4/CC. The Faradaic efficiency (FE) of CoP@CoMoO4/CC was determined by the ratio of measured to the theoretical H2 amount (Fig. S9), it showed nearly 100 % for HER.
The stabilities of CoP@CoMoO4/CC, CoP/CC, and CoMoP-800 were evaluated via a chronopotentiometry test at different current density, respectively. As illustrated in Figs. 6e and S8, the overpotential of CoP@CoMoO4/CC showed negligible increases of 0.6, 1.4, and 3.2 mV at current densities of 10, 30, and 50 mA cm-2, respectively. The potential of CoP/CC at 10 mA cm-2 drastically increased. Although the stability of CoMoP-800 is better than that of CoP, the potential of CoMoP-800 increased at the current density of 30 mA cm-2. We also characterized the electronic state and morphology of CoP@CoMoO4/CC after the stability test. As shown in Fig. S11, XPS indicated that the elemental species of CoP@CoMoO4/CC remained unchanged after HER. The SEM images (Fig. S12) revealed negligible corrosion on the surface of the sample, and the morphology has not collapsed or fallen off. However, the CoP nanowires (Fig. S13) have collapsed after the stability test, and the CoMoP-800 (Fig. S14) contained a large area of exposed CC, indicating that the material had fallen off. These results imply that the CoP and CoMoO4 heterostructures possess better electrochemical stability than pure phosphide CoP and CoMoP-800. This excellent electrochemical stability may be due to the optimal stability of the bimetal oxide and its heterostructures with metal phosphides. The CoP and CoMoO4 heterostructures would facilitate hydrogen escape, and the presence of oxides can be beneficial to maintaining catalyst morphology, further improving the HER catalytic performance. Based on Tafel slope, overpotential and stability, CoP@CoMoO4/CC heterostructures have excellent electrocatalytic properties compared with previously-reported CoP in recent years (Table 1).
Table 1 Comparison of HER performance for CoP@CoMoO4/CC with other HER electrocatalysts.
| Catalyst | Electrolyte | Tafel slope (mV dec-1) | η (mV) at j = 10 mA cm-2 | Stability test |
|---|---|---|---|---|
| CoP/CoMoO4@CC C-CoP-1/12 [35] | 1 M KOH 1 M KOH | 69 63 | 81 173 | 30 h 24 h |
| M-CoP/HPFS(Ni,Mn,Fe) [12] | 1 M KOH | 71-93 | 92-164 | 21 h |
| CoP aerogel [36] | 1 M KOH | 72 | 154 | 20 h |
| Co2P/Ti [37] | 0.5 M H2SO4 | 71 | 143 | 1000 cycle |
| CoP@3D-NPC [11] | 1 M KOH | 71 | 203 | 6 h |
| NiFeP@CoP/CC [38] | 1 M KOH | 71 | 96 | 24 h |
| Ce-CoP/CNT [39] | 0.5 M H2SO4 | 78 | 146 | 1000 cycle |
| CoP/CN@MoS2 [40] CoP@a-CoOx/C [41] | 1 M KOH 1 M KOH | 69 132 | 144 143 | N/A 24 h |
The rate of HER in the alkaline solution was determined by the Volmer step (H2O + e- + cat → Had-cat + OH-) [42]. Had-cat represents hydrogen adsorption at the active site of the catalyst. Another intermediate OH- produced by the Volmer step will prevent the active site of the catalyst from affecting the mutual binding of the subsequently adsorbed hydrogen. As we have previously reported, the heterostructure would strengthen hydrogen bonding and render ΔGH* close to 0 eV. Thus, at the interface between CoP and CoMoO4, the strong electronegativity of P can promote the adsorption of positively charged protons by CoP, making it a superior adsorption site for Had. The strong electrostatic affinity of Co2+ and Mo6+ in CoMoO4 is favorable for attracting negatively charged OH- [43]. The electronegativity-electrostatic affinity synergy between the CoP and CoMoO4 heterostructure interfaces accelerated Had through the catalyst in the Volmer step and the desorption of H in the Heyrovsky step, consequently accelerating HER kinetics in the alkaline solution.
4. Conclusion
We prepared a Co3O4@CoMoO4 heterostructure on a flexible conductive base (CC) via the hydrothermal method. We partially phosphatized the heterostructure through the CVD phosphating method based on the different stabilities of single- and double-metal oxides. CoP@CoMoO4/CC was successfully prepared. With an overpotential of 65 mV and Tafel slope of 69 mV dec-1 for HER, the excellent synergy between CoMoO4 and CoP is superior to that of most metal phosphide electrocatalysts. The selective attraction of H and OH- at the interface of the heterostructure considerably enhanced HER performance in alkaline solutions. In addition, the durability of the prepared CoP@CoMoO4/CC in 1 M KOH is superior to that of pure phosphide because of its combination with a bimetal oxide. Our present research provides a new method for preparing inexpensive, highly active, and highly durable catalysts for HER.
Declaration of Competing Interest
The authors declare no competing financial interest.
Acknowledgements
Our work was supported by the Key Research and Development Project of Hainan Province (No. ZDYF2018106), the National Natural Science Foundation of China (No. 51901059, No. 11563003, No. 11203009).
Appendix A. Supplementary data
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.jmst.2019.12.013.
Reference

Cobalt phosphide (CO2P) nanorods are found to exhibit efficient catalytic activity for the hydrogen evolution reaction (HER), with the overpotential required for the current density of 20 mA/cm(2) as small as 167 mV in acidic solution and 171 mV in basic solution. In addition, the Co2P nanorods can work stably in both acidic and basic solution during hydrogen production. This performance can be favorably compared to typical high efficient non-precious catalysts, and suggests the promising application potential of Co2P nanorods in the field of hydrogen production. The HER process follows a Volmer-Heyrovsky mechanism, and the rates of the discharge step and desorption step appear to be comparable during the HER process. The similarity of charged natures of Co and P in the Co2P nanorods to those of the hydride-acceptor and proton-acceptor in highly efficient Ni2P catalysts, [NiFe] hydrogenase, and its analogues implies that the HER catalytic activity of the Co2P nanorods might be correlated with the charged natures of Co and P. (C) 2014 Elsevier Ltd.

 WeChat
WeChat
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}