{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Multiple-Sized Amphiphilic Janus Gold Nanoparticles by Ligand Exchange at Toluene/Water Interface
[Kun Luo*  , Yongdong Xiang, Haiming Wang, Li Xiang, Zhihong Luo
, Yongdong Xiang, Haiming Wang, Li Xiang, Zhihong Luo* ]
, Yongdong Xiang, Haiming Wang, Li Xiang, Zhihong Luo]
|
|


Amphiphilic Janus gold nanoparticles (GNPs) are prospected to encapsulate drug molecules in cancer therapy and to serve as heterogeneous catalysts at oil/water interfaces, where Janus GNPs with different sizes are required. In this work, multiple-sized precursor GNPs were synthesized by seeded growth method protected with tris(hydroxymethyl)phosphine oxide (THPO) ligand molecule, and a ligand exchange reaction with triphenylphosphine (PPh3) at the toluene/water interface was employed to prepare amphiphilic Janus GNPs. UV-vis and transmission electron microscopy (TEM) analyses indicate that the as-prepared GNPs are nanocrystals with average diameters of 2.3 nm, 9.5 nm, 16.1 nm and 18.8 nm, respectively. Contact angle, Raman and X-ray photonic spectroscopy (XPS) analyses reveal that the self-assembled GNP films exhibit hydrophilic on one side and hydrophobic on another, owing to the adsorption of hydrophilic ligands (THPO and THP) and a similar amount of hydrophobic ligands (PPh3 and PPh3O). Angle-resolved XPS analysis further demonstrates that the individual GNPs actually possess hydrophilic and hydrophobic compartments on the surface, which regularly packed by supramolecular interactions at toluene/water interface to form the self-assembled GNP films.
Gold nanoparticles (GNPs) have been recognized as an attractive candidate for cancer therapy[1, 2, 3] and heterogeneous catalysis[4, 5], owing to their excellent biological compatibility, chemical stability as well as availability of morphological control and surface functionalization. These applications closely relate with the size of GNPs. As contrast agents, 1.9-nm GNPs have been used in vivo, demonstrating longer retention time and superior contrast to iodine with resolution of vessels as small as 100 µ m of heavily vascularized tumors [6]. In hyperthermia of cancer treatment, particles with 100-nm silica cores and a 15-nm GNP coating with resonance peak at 650-950 nm were reported to result in irreversible damage to SK-BR-3 human breast tumors under a tuned laser [7, 8]. As drug carrier, 5-nm GNPs covalently bound to cetuximab and gemcitabine were noticed to be superior to any of the agents alone or in combination in vitro and in vivo in pancreatic cancer [9]. In another study, the size of 40-50 nm was suggested to be optimal for the GNPs bound with trastuzumab antibody in human SK-BR-3 breast cancer. Besides, the GNPs can also encapsulate the drug molecules in form of submicronic colloids [2, 3], which can be released under control at the target tumor cells[10]. Small GNPs allow more payload in the “ microcapsules, ” but they suffer from unstable adsorption at oil/water interface, because the desorption energy of homogeneous GNPs relies heavily on the contact angle when they are smaller than 10 nm[11]. In contrast, the energy of amphiphilic Janus particles that exhibit two distinguished surface chemistries on the two sides[12], may be increased 3-fold by maximizing the amphiphilicity[13], which behave analogue to conventional surfactant molecules[14] and are able to serve as Pickering emulsion stabilizer. The amphiphilic Janus GNPs microcapsules are also prospective to play directly as contrast and hyperthermia agents.
Synthesis of amphiphilic Janus nanoparticles requires creating two distinct compartments on the surface in a cost-effective and reliable way. Masking was one of the techniques initially applied on larger Janus particles and scaled down to the nanoscale, involving the protection of one side and modification of another, followed with the removal of the protection. However, the use of gas/liquid, liquid/liquid, and liquid/solid interfaces that trap homogeneous nanoparticles is much more popular. Pradhan et al.[15, 16] compressed the hexane- thiolate-protected GNPs (ca. 2 nm) at water surface by Langmuir method and removed the film on a glass slide, and then was immersed into a 2-(2-mercaptoethoxy)ethanol aqueous solution, resulted in amphiphilic Janus GNPs. Similar approach was adopted by Xu et al.[17] to prepare biphasic Janus GNPs with other S-containing molecules. Andala et al.[18] made use of toluene/water emulsion interface to trigger the ligand exchange reaction of dodecanethiol and mercaptoundecanoic acid from both phases, to replace the protection molecule of the precursor dodecylamine-protected GNPs, led to the amphiphilic Janus GNPs (9.2 nm). Granick et al.[19] employed molten paraffin wax instead of the oil, to immobilize silica NPs as the wax was cooled, leaving the half in water available for the modification with (aminopropyl)triethoxy- silane. Janus particles were then obtained as the wax was dissolved with chloroform. Other than surface chemistry, the control over particle size and geometry are also taken into account on the synthesis of Janus particles. For example, the use of polymeric stabilizers led to uniform GNPs with difference sizes[20, 21], and Au nanowires[22] and Au-M bimetallic nanoparticles[23] were also prepared for surface modification. However, there are fewer studies on the synthesis and characterization of the amphiphilic Janus GNPs with ligands other than S-containing molecules, which are equally required on chemical and medical innovations. In this work, multiple-sized amphiphilic Janus GNPs modified with P-containing molecules were prepared, by means of ligand exchange from homogeneous precursor GNPs at toluene/water interfaces, and the surface property of the as-prepared GNPs was also investigated.
Chloroauric acid (HAuCl4, 47.8%, AR) was purchased from Shanghai Billiton Co. Ltd., China. Triphenylphosphine (PPh3, CP) and toluene (99.5%, AR) were bought from Sinopharm Chemical Reagent Co. Ltd., China. Tetrakis(hydroxymethyl)phosphonium chloride (THPC, 80%, CP) was from Tokyo Chemical Industry Co. Ltd., Japan. Sodium hydroxide (NaOH, 96%, AR) and hydroxylamine hydrochloride (96%, AR) were purchased from Shantou Xilong Chemical Co. Ltd., China. Trisphosphine oxide (THPO, 95.5%) was bought from Wuhan Jinlin Chemical Technology Co. Ltd., China. Deionized water (18.2 MΩ cm) was employed to prepare the solutions in the experiments.
NaOH (1 mol/L, 300 µ L), THPC (50 mmol/L, 1 mL) and HAuCl4 (25 mmol/L, 2 mL) were sequentially added to a conical flask containing 47 mL deionized water under ultrasonic stirring, leading to the formation of a GNPs colloid in palm red color, which were removed in a dialysis bag (with intercept molecular weight of 3500) immersed in deionized water to remove soluble reaction products. The operation lasted for 6-7 days accompanied with the refreshment of deionized water periodically, resulted in the pure GNP colloid marked as Colloid A.
The preparation of larger GNPs was carried out by seed growth method in sequence. First, 1 mL of 25 mmol/L HAuCl4, 2 mL of 50 mmol/L THPO and 500 µ L of 200 mmol/L hydroxylamine hydrochloride were added in 80 mL deionized water under ultrasonic stirring, followed with the addition of 10 mL of Colloid A, leading to the pure Colloid B after dialysis treatment. Second, 10 mL of Colloid B was added into the fresh solution containing 1 mL of 25 mmol/L HAuCl4, 2 mL of 50 mmol/L THPO and 375 µ L of 200 mmol/L hydroxylamine hydrochloride, resulted in the pure Colloid C after dialysis treatment. Finally, 10 mL of Colloid C was added into the fresh solution containing 1 mL of 25 mmol/L HAuCl4, 2 mL of 50 mmol/L THPO, and 250 µ L of 200 mmol/L hydroxylamine hydrochloride resulted in the pure Colloid D after dialysis treatment.
Surface modification of the GNPs in the as-prepared colloids was carried out at the toluene/water interface by ligand exchange reaction. 5 mL of the as-synthesized colloids (Colloid A, B, C, and D) were added into four vials, in which 5 mL of 50 mmol/L PPh3 toluene solutions were added, respectively. Then, the lids of the vials were made close, and violent shaking was applied to the vials for 10 min, leading to the presence of the self-assembled GNP films at the toluene/water interface after standing for 12 h, similar to Binks et al.[24]. These films were lifted up with small glass slides placed in water underneath the interface, leaving the hydrophobic sides upwards. Then, the films were rinsed separately with acetone and deionized water for three times, which were dried at ambient temperature and named as film A, B, C, and D, in correspondence with the precursor Colloid A, B, C, and D.
The morphology of the GNPs was characterized by transmittance electron microscopy (TEM, JEM-2010, JEOL) operated at 200 kV. UV-vis absorption spectrum (Beijing Purkinje General Instrument Co. Ltd.) was used to characterize the as-synthesized GNPs. Confocal Raman microscopy (DXR, Thermo Scientific) equipped with a laser of 532 nm, X-ray photonic spectroscopy (XPS, AXIS ULTRA XPS, KRATOS) were employed to determine the capping molecules of the as-prepared GNPs. A contact angle analyzer (JWA-360A, Xiamen Congda Smart Technology Co. Ltd., China) was used to measure the surface wettability of the GNP assemblies, where the hydrophobic side of a film on glass substrate was measured directly with a drop of 5 µ L deionized water. As for the hydrophilic side, another slide with a piece of double-sided adhesive tape was overlain onto the rinsed film, leading to the overturned film available for contact angle measurement.
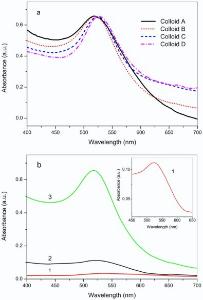
The UV-vis absorbance spectra of Colloid A, B, C, and D are shown in Fig. 1(a), in which red shifts are noticed for the absorbance peaks of the Colloid A, B, C, and D in sequence, suggesting the increase of particle size of the GNPs[25]. Compared to the THPC solution, the ability of hydroxylamine hydrochloride seemed to be weaker on the reduction of HAuCl4, leading to a low yield of GNPs in line with the previous literature[26]. As displayed inFig. 1(b), a weak UV-vis absorbance appears at 517 nm (curve a, enlarged in the inset) when 375 µ L of 200 mmol/L hydroxylamine hydrochloride were used to reduce HAuCl4 with the presence of THPO as capping molecule. On the other hand, THPC can act as reducing agent to generate THPO-protected GNPs directly, and the absorbance in curve 2 appears stronger than curve 1, indicative of a larger concentration of GNPs obtained. However, the reduction of hydroxyl- amine hydrochloride was apparently accelerated as Colloid A was added as seed for the GNP growth, ending up with the largest absorbance at 523 nm in curve 3, which is even bigger than the sum of the curve 1 and 2. Similar phenomenon was also observed in the synthesis of the Colloid C and D (not shown), probably due to the lower energy needed for grain growth than for nucleation.
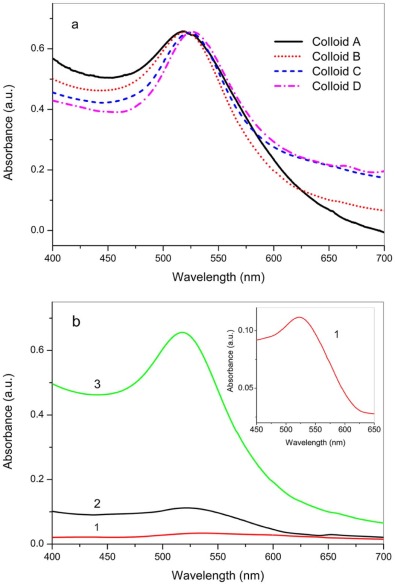 | Fig. 1. UV-vis spectra of the as-synthesized colloids: (a) UV-vis absorbance of Colloid A, B, C, and D; (b) the effect of seeded growth of GNPs, curve 1 shows the absorbance with hydroxylamine hydrochloride as reducing agent in HAuCl4 solution, curve 2 is the absorbance with THPC, and curve 3 shows the reduction by hydroxylamine hydrochloride with addition of Colloid A. Inset: enlarged absorbance peak of curve 1. |
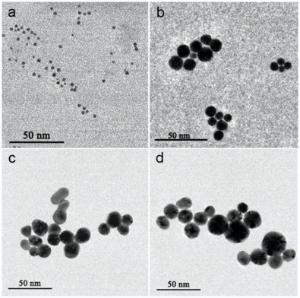
The GNPs in the as-synthesized colloids were examined by TEM analysis. As presented inFig. 2(a), the GNPs in Colloid A appear uniform in morphology and particle size (2.3 nm in average diameter). In contrast, the GNPs in Colloid B, C, and D (as shown in Fig. 2(b-d)) become much larger after seeded growth, which are 9.5 nm, 16.1 nm, and 18.8 nm in average diameter, respectively, in agreement with the UV-vis results. However, the morphology of individual GNPs in the Colloid B, C, and D vary considerably.
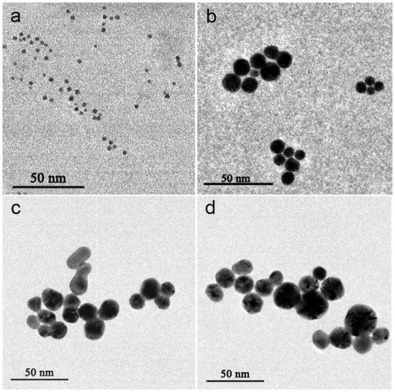 | Fig. 2. TEM images of GNPs obtained from Colloid A (a), Colloid B (b), Colloid C (c), and Colloid D (d). |
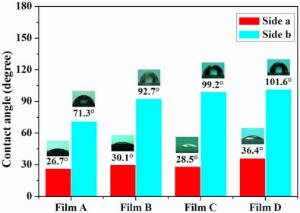
The toluene/water interface was employed to modify the as-prepared homogenous GNPs with different sizes, and the ligand exchange reaction between the THPO-protected homogeneous GNPs and PPh3 in toluene was carried out at the interface, resulted in hydrophilic and hydrophobic compartments on the GNP's surface due to the confinement of the immiscible interface for the PPh3 molecule. To offer more contact opportunities, strong handshaking was applied to generate a large amount of emulsion drops, which coalesced to form a self-assembled GNP film after standing for a period of time, to allow ligand exchange at the toluene/water interface. It is difficult to directly analyze the surface property of individual GNPs, but the contact angle measurement is available for the surface characterization of regularly assembled GNP films. As shown in Fig. 3, all the four self-assembled GNP films of precursor Colloid A, B, C, and D exhibit wettability difference on both sides after interfacial ligand exchange, i.e. 26.7° , 30.1° , 28.5° , and 36.4° at one side, and 71.3° , 92.7° , 99.2° , and 101.6° on the other, respectively. It is noticed that the contact angles on the hydrophobic sides of the self-assembly films increase with the size of GNPs, probably due to the less regular surface containing uncoalesced “ microdroplets” covered by the small Janus GNPs, which was not present for the larger GNPs (> 10 nm) by the confinement of the toluene/water interface. The fluctuating contact angles on the hydrophilic sides possibly relate with the interference from the background adhesive tapes, which was used to overturn the thin film assemblies.
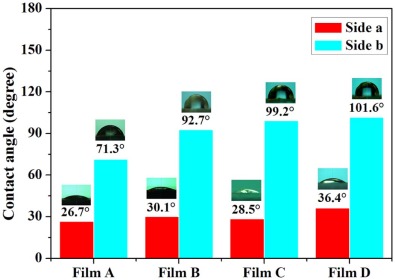 | Fig. 3. Contact angle of water on the hydrophilic (Side a) and hydrophobic surface (Side b) of films A, B, C, and D. |
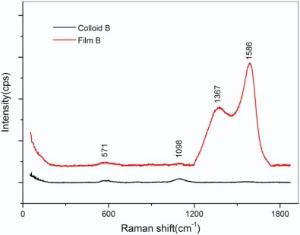
Raman spectroscopy was employed to differentiate the homogeneous and Janus GNPs, where the GNPs in Colloid B and the film B after ligand exchange reaction were compared. As illustrated in Fig. 4, the film B presents four peaks, and the two shoulders at 571 cm-1and 1098 cm-1 can be attributed to C-P and P=O vibration of THPO and THP, while the two larger peaks located at 1586 cm-1 and 1367 cm-1 are assigned to the vibration of benzene belonging to PPh3 or PPh3O. The result suggests that the co-existence of THPO (THP) and PPh3 (PPh3O) in the self-assembled film B. In contrast, only two Raman peaks are seen at 571 cm-1 and 1098 cm-1 for the Colloid B, indicative of the homogeneous GNPs protected by THPO and THP ligands.
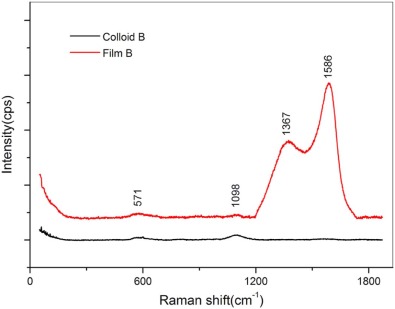 | Fig. 4. Raman spectra of Colloid B and film B after ligand exchange. |
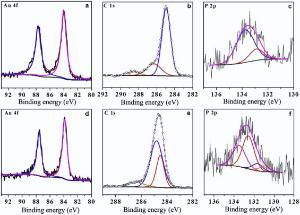
Fig. 5 shows the XPS survey of the GNPs in the Colloid B and film B. Two fitted peaks at 84.4 eV and 87.6 eV in Fig. 5(a) and (d) can be assigned to the Au4f5/2 and Au4f7/2[27], indicative of metallic gold. In Fig. 5(b), three fitted peaks for the C1s signal at 285.0 eV, 286.3 eV and 288.7 eV belong to — CH2OH[28], C=O and O— C=O[29] groups, respectively. The fitting of the P2p curve shown in Fig. 5(c) results in two peaks at 133.8 eV and 132.9 eV, corresponding to THPO[30] and THP, respectively. Hence, only hydrophilic ligand THPO or THP is present on the GNPs in Colloid B. In Fig. 5(e), three fitted peaks available for the C1s signal at 284.6 eV, 285.0 eV, and 286.6 eV are assigned to — CH2OH, (C6H5)3PO, and C
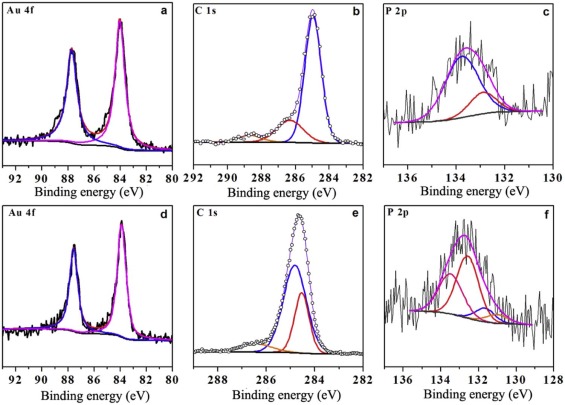 | Fig. 5. XPS spectra of Au 4f, C1s, and P 2p obtained from Colloid B (a, b, c) and self-assembled film B (d, e, f). |
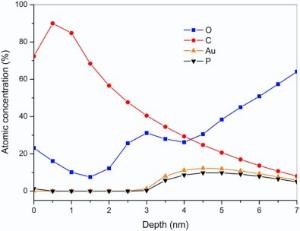
Angle resolved X-ray photoelectron spectroscopy (ARXPS) further provides with a detailed in-depth profile for the self-assembled GNP films, where the element distribution in depth can be calculated by the maximum entropy (MaxEnt) method. As shown in Fig. 6, C content increases from 72.4% to 90%, and P decreases from 1.3% to 0, and O reduces from 23.1% to 10.1% in the depth range from 0 nm to 1 nm, corresponding to toluene solvation layer with small amount of PPh3O, originated from the oxidation of PPh3. As the depth varies from 1.5 nm to 3.0 nm, the C content declines from 68.4% to 40.5%, O content rises up from 7.5% to 31.2%, and the others remain less than 0.3%, corresponding to an acetone solvation layer with PPh3 and THPO, possibly introduced in the rinsing process. From 3.5 nm to 4.5 nm, the content of C declines from 34.5% to 24.8%, but the content of O increases from 27.9% to 30.6%, Au from 8.0% to 12.3%, and P from 5.8% to 9.8%, implying the existence of PPh3O and PPh3. From 4.5 nm to 5.5 nm, the C keeps decreasing to 17.0%, Au to 10.9%, and P to 9.1%, but the content of O continues rising to 45.0%, suggesting the appearance of hydrophilic ligands of THPO and THP. As the depth exceeds 6.0 nm, the background noise of O from the quartz substrate overwhelms the spectra of Au, P, and C. However, the biphasic distribution of PPh3O and PPh3 against THPO and THP on the first layer of the GNP self-assembly is examined. Considering the randomness on the distribution of the GNPs during the emulsion coalescence, the other GNPs after interfacial ligand exchange can also be considered to have the same biphasic surface chemistry as those assembled in the first layer.
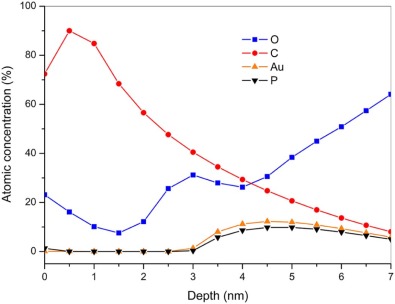 | Fig. 6. In-depth distribution of O, C, Au, and P elements in the self-assembled film B. |
A series of GNPs were synthesized prepared by seeded growth with average diameters of 2.3 nm, 9.5 nm, 16.1 nm, and 18.8 nm, according to UV-vis and TEM analyses. Ligand exchange reaction was triggered as the precursor homogeneous GNPs were in contact with the PPh3 in toluene, leading to thin film assemblies of GNPs at the toluene/water interface, which present hydrophobic property on one side and hydrophilic on another according to the contact angle measurement. Raman and XPS results indicate that both hydrophilic and hydrophobic groups are examined on the GNPs in film B, where the atomic ratio of PPh3 and PPh3O to THPO and THP is about 1.3. ARXPS analysis further demonstrates that the hydrophobic (PPh3 and PPh3O) and hydrophilic (THPO and THP) ligands actually distribute on different halves of the individual GNP's surface, leading to amphiphilic Janus GNPs. This facile protocol to synthesize multiple-sized amphiphilic Janus GNPs is prospected to meet requirements for the chemical and medical innovations.
This work was supported by the National Natural Science Foundation of China (No.21163004) and Guangxi Natural Science Foundation (Nos. 2015GXNSFBA139220 and2013GX NSFAA019029).
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|