





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Facile Synthesis of Bimetallic Au@Pd Nanoparticles with Core-shell Structures on Graphene Nanosheets
[Nanting Li, Shaochun Tang, Xiangkang Meng*  ]
]
]
|
|


In this paper, we report a simple one-step thermal reducing method for synthesis of bimetallic Au@Pd nanoparticles with core-shell structures on the graphene surface. This new type of Au@Pd-G composites is characterized by transmission electron microscopy, high resolution transmission electron microscopy, X-ray photoelectron spectroscopy and X-ray diffraction. It is found that Au@Pd nanoparticles with an average diameter of 11 nm are well dispersed on the graphene surface, and the Au core quantity as well as the Pd shell thickness can be quantitatively controlled by loading different amounts of metallic precursors, and the involved core-shell structure formation mechanism is also discussed. The ternary Pt/Au@Pd-G composites can also be synthetized by the subsequent Pt doping. The catalytic performance of Au@Pd-G composites toward methanol electro-oxidation in acidic media is investigated. The results show that Au@Pd-G composites exhibit higher catalytic activity, better stability and stronger tolerance to CO poisoning than Pd-G and Au-G counterparts.
Direct methanol fuel cells (DMFCs) are the promising candidates for power sources of portable electronic devices and fuel cell vehicles due to the much higher energy density of liquid methanol fuel than gaseous fuels such as hydrogen[1]. As compared with other catalysts, platinum (Pt)-based catalysts are still the most effective catalysts in DMFCs[2]. However, the high cost, low availability and low tolerance to CO poisoning of Pt-based catalysts limit their applications in DMFCs[3] and [4].
The use of palladium (Pd) in fuel cell catalysts[5] is interesting, because it is more widespread in the earth crust than Pt (abundance of 1.5 × 10− 2vs. 5 × 10− 3 parts per million by mass, respectively), and it is less expensive than Pt. Replacing Pt by Pd will decrease the electrodes' cost [6] and [7]. In addition, Pd is very stable in the acidic fuel cell environment [8] and also exhibits interesting electro-catalytic properties[9], [10], [11] and [12]. These properties have led to consider Pd as a possible catalyst for these reactions, such as methanol, ethanol [13], [14] and [15] and formic acid [16], [17], [18] and [19] electro-oxidation in which CO appears as a poisoning intermediate.
Recently, Pd-M bimetallic nanoparticles (NPs) have received more and more attention as electro-catalysts[20]. Moreover, bimetallic NPs with a unique core-shell structure can minimize the use of precious metal precursors and are expected to show enhanced catalytic activity and selectivity[21], [22] and [23]. Considering that Au (electronegativity χ = 2.4) is the transition metal which is more electro-negative than Pd (χ = 2.2), the incorporation of Au has unique effects on Pd NPs. Therefore, bimetallic Pd-Au NPs are very important as fuel cell catalysts, especially for methanol electro-oxidation[24] and [25].
To maximize the electro-catalytic activity of Pd-Au NPs, a suitable carbon support is required to disperse these NPs. Graphene, a single-atom-thick sheet of hexagonally arrayed sp2-bonded carbon atoms, has been extensively studied in physics, chemistry, and material fields[26] and [27]. Due to its huge surface area (~2600 m2 g− 1), high electrical conductivity (105-106 S m− 1), and excellent catalytic activity[28] and [29], graphene has been considered as a promising candidate used for a new 2D support to load Pt, Pd, and Au NPs for applications in fuel cells[30], [31], [32] and [33].
Hence, we report a new type of Au@Pd core-shell structured NPs-graphene (Au@Pd-G) composites synthetized by means of a simple one-step method, in which PdCl2 and HAuCl4 are reduced simultaneously by ascorbic acid in the presence of the complexing agent of trisodium citrate. The products are characterized by transmission electron microscopy (TEM), high resolution TEM (HRTEM), X-ray photoelectron spectroscopy (XPS) and X-ray diffraction (XRD). The results reveal that Au@Pd NPs with an average diameter of 11 nm are well dispersed on the graphene surface, and the Au core quantity as well as the Pd shell thickness can be quantitatively controlled by loading different amounts of metallic precursors, and the involved core-shell structure formation mechanism is also discussed. The ternary Pt/Au@Pd-G composites can also be synthetized by the subsequent Pt doping. Au@Pd-G composites exhibit higher catalytic activity, better stability and stronger tolerance to CO poisoning toward methanol electro-oxidation in acidic media for DMFCs in comparison with Pd-G and Au-G counterparts.
Natural graphite powder (spectrum pure), H2SO4 (98 wt%, A.R.), KMnO4 (A.R.), H2O2 (30 wt%, A.R.), HCl (36-38 wt%, A.R.), NaOH (A.R.), KOH (A.R.), BaCl2 (C.P.), ascorbic acid (A.R.) and anhydrous ethanol were all purchased from Sinopharm Chemical Reagent Co. Ltd. Trisodium citrate was obtained from Shanghai Precise Analysis Chemical Technology Co. Ltd, and methanol was obtained from Nanjing Chemical Reagent Co. Ltd. PdCl2, HAuCl4 and K2PtCl6 were all purchased from Nanjing Ning-Shi Chemical Reagent Co. Ltd, and ethylene glycol (EG) was obtained from Shanghai Jiu-Yi Chemical Reagent Co. Ltd, finally monosodium glutamate was purchased from a supermarket. All reagents were used as received without further purification. Deionized water with a resistivity of exceeding 18.0 MΩ cm from a JL-RO 100 Millipore-Q Plus water purifier was used throughout the experiments.
Graphite oxide was synthetized from natural graphite powder using the modified Hummers' method[34]. Typically, 10 g of graphite powders was added into 230 mL of 98 wt% H2SO4, followed by an intense stirring for 5 min at room temperature, then 30 g of KMnO4 was introduced into the mixture. Under continuously vigorous stirring, the mixture was kept at the low (10-15 ° C), middle (35 ± 3 ° C) and high (98 ° C) temperatures for 2 h, 30 min, and 30 min reactive stages, respectively. Subsequently, 230 mL of diluted H2O2 (5 wt%) was added into the above mixture. Finally, the suspension was vacuum filtrated using 460 mL of deionized water and 300 mL of diluted HCl (5 wt%). BaCl2 aqueous solution was used to test whether the filtration solution contained SO42− , and NaOH (5 mol L) was added to produce flocculation. The resulting products were centrifuged, followed by drying at 50 ° C in an oven.
Graphite oxide was exfoliated into graphene oxide (GO) by sonication in water[35]. The typical procedure as follows: excessive trisodium citrate (∼0.27 g) was added to 45 mL GO suspension (0.67 mg/mL) and sonicated for 30 min. Then 1.6 mL (24.3 mmol/L) PdCl2, 494.6 μ L (24.3 mmol/L) HAuCl4 and 2.3 mL (0.1 mol/L) excessive ascorbic acid (AA) were added subsequently. The mixing solution was heated at 100 ° C in an oil bath with mechanical stirring for 2.5 h. After cooling down to room temperature, the products (Au@Pd-G composites) were washed extensively with anhydrous ethanol and deionized water, then centrifuged several times, finally dried at 80 ° C in a vacuum oven. All other conditions were unchanged, and the composites synthesized using 1.6 and 2.0 mL PdCl2 were defined as Au@Pd-G(1) and Au@Pd-G(2), respectively. The composites synthesized using 1.6 mL PdCl2 and 550.6 μ L HAuCl4were defined as Au@Pd-G(3). The procedures for synthesis of monometallic Pd-G and Au-G composites were the same as above, but only 1.6 mL PdCl2 or 494.6 μ L HAuCl4 was used.
The obtained Au@Pd-G(1) composites (0.008 g), 1.2 mL K2PtCl6 (7.88 mmol/L) and 100 mg monosodium glutamate were mixed together in 40 mL KOH/EG at a given pH of ca. 13. Subsequently, the solution was ultrasonicated and magnetically stirred vigorously each for 30 min to obtain a homogeneous suspension. Upon completion, the suspension was transferred into a 50 mL Teflon-lined stainless-steel autoclave. The autoclave was sealed, heated at 160 ° C for 3 h, and air-cooled to room temperature. The obtained resulting products denoted as Pt/Au@Pd-G(1) were washed and centrifuged with deionized water several times. Finally, the lyophilization was applied to prevent the aggregation of graphene sheets during the drying process.
The size, distribution, and morphology of the products were characterized on TEM, HRTEM images and selected area electron diffraction (SAED) patterns were obtained on a JEOL JEM-2100 TEM operating at 200 kV. XPS was measured on a Thermofish K-alpha X-ray photoelectron spectrometer. Binding energies were determined with reference to carbon's C1s of 284.6 eV. The products' composition and crystallographic properties were analyzed by XRD on a Rigaku Ultima III diffractometer (using Cu Kα of 0.15418 nm radiation) at a scanning rate of 2° min− 1 in the range of 5° -90° .
Electrochemical measurements were performed with a CHI 660D electrochemical analyzer (CH Instruments, Inc., Shanghai). A conventional three-electrode cell was used, including a glassy carbon electrode (GCE, geometric area of 0.1256 cm2) as the working electrode, a saturated calomel electrode (SCE) as the reference electrode and a platinum foil as the counter electrode. The potentials were measured with SCE as the reference.
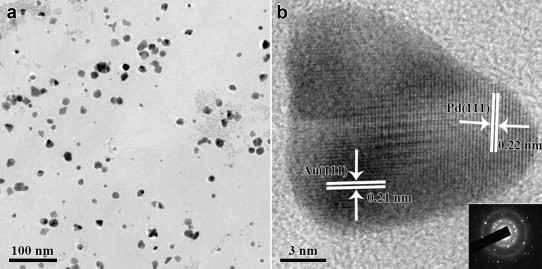
Fig. 1(a) is the TEM image of Au@Pd NPs on graphene nanosheets, and NPs are well dispersed on the graphene surface which is not a flat plane with surface wrinkles being clearly observed. As shown inFig. 1(b), the red-brownish GO solution turns dark when the reduction is complete. The Pd and Au elements co-exist on the graphene surface, and the supported NPs show a dark core (Au) and a comparatively pale shell (Pd) from HRTEM (Fig. 1(c)), indicating the formation of bimetallic Au@Pd core-shell NPs on the graphene surface[36]. Fig. 1(c) also shows the typical gold core's lattice with a d-spacing of 0.21 nm, corresponding to (111) planes of face-centered cubic (fcc) Au [37], while the shell'sd-spacing is 0.22 nm, corresponding to (111) planes of fcc Pd [38]. Inset 1(c) is the corresponding SAED pattern of an individual core-shell NP in Fig. 1(c), confirming the core-shell NP's monocrystalline nature.
 | Fig. 1 (a) TEM image of Au@Pd nanoparticles on graphene nanosheets; (b) a comparison of the solution before and after the reaction; (c) HRTEM image of an individual core-shell NP of Au@Pd-G(1) and the corresponding SAED. |
shows TEM images of Au@Pd-G(3) using 1.6 mL PdCl2 and 550.6 μ L HAuCl4. The polygonal Au@Pd core-shell NPs are evenly distributed on the graphene surface in Fig. 2(a), showing the existence of triangular, spherical etc. Au@Pd core-shell NPs on the graphene surface with increasing the Au content. Fig. 2(b) is the high-magnified TEM image of a single triangular Au@Pd NP, and the inset 2(b) is the corresponding SAED pattern, confirming the monocrystalline nature.
 | Fig. 2 (a) TEM image of Au@Pd-G(3) shows the existence of triangular, spherical etc. Au@Pd core-shell NPs on the graphene surface; (b) high-magnified TEM image of a single triangular Au@Pd NP and the corresponding SAED pattern. |
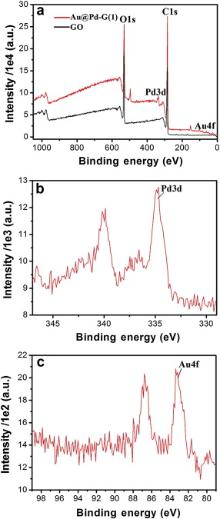
Fig. 3(a) shows XPS patterns of GO and Au@Pd-G(1) before and after the reduction. The bands at 284.8 and 531.0 eV are associated with C1s and O1s, respectively, and the oxygen-containing functional groups' partial removal contributes to the composites' stability in water as shown in Fig. 1(b). Fig. 3(b) is the typical Pd3d XPS pattern of Au@Pd-G(1), showing that composites mainly consist of Pd0 with a small amount of Pd2+ at 336.1 and 341.2 eV which may be caused by the surface oxidation during the synthesis process. By calculating the peak area, the Pd content is 0.41 wt% and the Au content is 0.07 wt%. Given that XPS can only penetrate surfaces' several nanometers, this result strongly supports the formation of Au@Pd core-shell structures, as the signal from the Au core can be significantly shielded by the Pd shell in XPS analysis, which leads to the significantly low Au content with strong noises from the base line. In addition, the appearance of Au4f at 83.8 and 87.6 eV in Fig. 3(c) confirms the complete reduction of Au3+ to Au0.
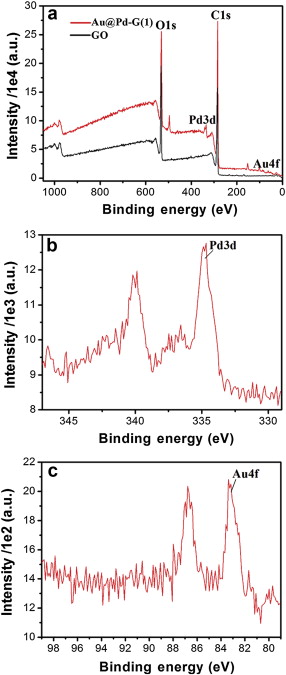 | Fig. 3 XPS patterns of (a) GO and Au@Pd-G(1), (b) Pd3d and (c) Au4f. |
shows XRD patterns of GO, Au-G, Pd-G and Au@Pd-G(1)-(2). The presence of the diffraction peak at 11.2° (red curve) corresponding to the (002) inter-planar spacing of GO (0.791 nm) is due to the introduction of various oxygen-containing functional groups. When Au or Pd is supported on the graphene surface, patterns have broad diffraction peaks centered at 25° (pink and orange curves), confirming that the graphitic nature is restored by the reduction with ascorbic acid. XRD patterns of Au@Pd-G(1)-(2) (crimson and navy curves) reveal that the fcc structure of the NPs has distinguished diffraction peaks especially in (220) and (311) planes. While diffraction peaks of (111) and (200) planes in Au and Pd overlap with each other, it is difficult to distinguish them (the lattice mismatch factor for Pd-Au is about 4%)[39]. In addition, the peak intensity of the (111) plane in Au@Pd-G composites gradually becomes stronger with increasing the Pd content (the Pd shell thickness: Au@Pd-G(2) > Au@Pd-G(1)).
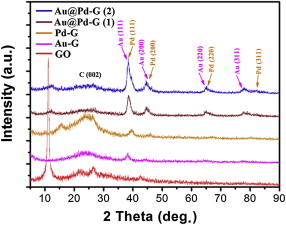 | Fig. 4 XRD patterns of GO, Au-G, Pd-G and Au@Pd-G(1)-(2). |
As for the core-shell structure formation of the bimetallic Au@Pd NPs on the graphene surface, we suggest a citrate-complexation-assisted controlled reducing mechanism. The trisodium citrate plays a key role in the formation of the bimetallic Au@Pd core-shell NPs. The reduction potential of AuCl4− /Au0and PdCl42− /Pd0vs RHE (reversible hydrogen electrode) is 1.02 and 0.6 V, respectively. The citrate anion can further reduce the reduction potential of PdCl42− /Pd0 by the complexation reaction with Pd2+. As a consequence, HAuCl4 and PdCl2 are reduced in order by ascorbic acid, and the following is details of the mechanism.
(1) At the initial stage, oxygen-containing functional groups on the GO surface serve as locating sites for AuCl4− ions which are reduced by excessive ascorbic acid, forming many small Au nuclei, and the GO is reduced to graphene simultaneously. (2) A spontaneous growth of Au NPs occurs. Due to oil bath heating, the graphene surface is very hot, resulting in that a spontaneous NPs growth happens selectively on the graphene surface, especially on the pre-existing Au nuclei, but not in the solution instead. The addition of Au NPs has the same rate in all directions, leading to the formation of the spherical-like Au core due to the surface energy minimization, and increasing the concentration of AuCl4− ions will increase the number of Au nuclei on the graphene surface. (3) After the reduction of HAuCl4 finishs, PdCl2 begins to be reduced by ascorbic acid outside the Au core, forming bimetallic Au@Pd NPs with core-shell structures on the graphene surface.
As for the growth mechanism of the polygonal Au@Pd-G(3) NPs, using 1.6 mL PdCl2 and 550.6 μ L HAuCl4 in Fig. 2, with increasing the Au content, Au cores are easy to form triangular, cubic, octahedral, decahedral, icosahedral and tetrahexahedral structures, which are described in detail in literature[40]. The Pd-shell component is overgrown on the (111) plane of the Au core, forming the aggregated polygonal Au@Pd NPs with core-shell structures on the graphene surface.
The ternary Pt/Au@Pd-G(1) composites can be synthesized by the solvothermal process, in which K2PtCl6 is selectively reduced on the Pd shell of Au@Pd-; G(1) by EG in the presence of monosodium glutamate. The formation of Pt/Au@Pd-G(1) is confirmed by TEM images in Fig. 5. Under the solvothermal process, EG will effectively reduce Pt ions and the existence of monosodium glutamate has an important effect on the nucleation and growth of Pt. As shown in Fig. 5(a), in the presence of monosodium glutamate, Pt NPs preferentially deposit on the Pd shell of Au@Pd-G(1) to form Pt/Au@Pd-G(1) which is still firmly anchored onto the graphene surface even after the ultrasonication for preparation of TEM sample, and the graphene surface has no redundant Pt NPs, while Pt NPs will non-selectively deposit on both the Pd shell of Au@Pd-G(1) and the graphene surface in the absence of glutamate (Fig. 5(c)). The HRTEM image in Fig. 5(b) further reveals the coexistence of lattice spacing of 0.22 nm and 0.23 nm on the Pd shell of Au@Pd-G(1), corresponding to the (111) inter-planar distance of the fcc Pd shell and the standard Pt crystalline lattice, respectively. These results show the hybrid structure of Pt and Pd in Pt/Au@Pd-G(1), and inset of Fig. 5(b) confirms the monocrystalline nature of Pt/Au@Pd-G(1). Fig. 5(d) is the corresponding HRTEM image of Fig. 5(c) without monosodium glutamate. Many small Pt NPs of 4-6 nm with different orientations are easy to aggregate on the graphene surface, and inset of Fig. 5(d) is the corresponding SAED pattern, confirming the fcc Pt NPs' polycrystalline nature.
 | Fig. 5 TEM images of Pt/Au@Pd-G(1) (a) with and (c) without monosodium glutamate; (b) and (d) are the corresponding HRTEM images of (a) and (c), respectively; Inset (b) and (d) are the corresponding SAED patterns. |
Fig. 6(a) shows the XPS pattern of Pt/Au@Pd-G(1) after the reduction. Besides the bands of C1s, O1s, Pd3d and Au4f, two peaks at 71.0 and 74.4 eV and the other two peaks at 314.8 and 330.1 eV can be assigned to Pt4f and Pt4d, respectively. Fig. 6(b) shows the doublet Pt4f7/2 and Pt4f5/2 peaks at binding energies of 71.0 and 74.4 eV, respectively. This confirms that Pt is present as Pt0 in Pt/Au@Pd-G(1) rather than a compound. By calculating of the peak area, the Pt content is 0.32 wt%.
 | Fig. 6 XPS patterns of (a) Pt/Au@Pd-G(1) and (b) Pt4f. |
The ionization reaction of glutamic acid in the solution can occur and the glutamate ions form, thus obtaining the negatively charged hydroxy by dehydrogenation and the positively charged amino by hydrogenation, as shown in the glutamate ion's molecular structure diagram of Fig. 7. The glutamate ion can be easily adsorbed on (111) planes of the Pd shell by the physical electrostatic adsorption. Due to that the outer five d-orbitals of Pt2+ has been filled by only eight electrons, leaving an empty d-orbital unfilled, the sole pair electrons on the negatively charged hydroxy (O− ) can coordinate with the empty d-orbital of Pt2+, leading to the deposition of Pt NPs on the Pd shell of Au@Pd-G(1) rather than the graphene surface.
 | Fig. 7 Glutamate ion′s molecular structure diagram and the outer electron distribution of O2− and Pt2+ can be used to explain why the selective deposition of Pt NPs can occur on the Pd shell of Au@Pd-G(1) rather than the graphene surface. |
It is reported that the NPs may go through a serious aggregation in the absence of supports[41], and the core-shell NPs without supports usually have larger diameters (20-50 nm)[42]. However, the introduced graphene nanosheets provide an excellent platform to stabilize bimetallic Au@Pd NPs with the diameter of about 11 nm which show a superior catalytic performance. Reactants e.g. methanol can easily access bimetallic Au@Pd NPs on both sides of the 2D graphene nanosheets which have a greater surface-to-volume ratio without limits, and diminishing the apparent contact time of products with catalysts can lead to more active and more selective catalytic effects.
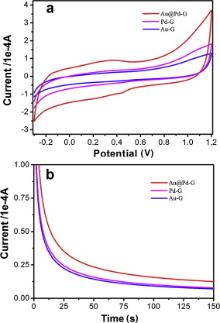
We evaluate the catalytic performance of Au@Pd-G, Pd-G and Au-G composites toward methanol electro-oxidation in acidic media under the same metal loading by CV and I-t measurements. As shown in Fig. 8(a), the CV area of Au@Pd-G is apparently larger than those of Pd-G and Au-G, indicating that Au@Pd-G composites have higher catalytic activity, better stability and stronger tolerance to CO poisoning than Pd-G and Au-G counterparts for methanol electro-oxidation. For I-t measurements, though all electrodes display an initial fast current decay followed by a slower attenuation, gradually reaching a pseudo-steady state, the Au@Pd-G electrode exhibits the highest initial current, and the current maintains the highest steady state during the whole measurement. This can be attributed to that the unique core-shell structure ensures the maximum contact of Pd to reactants [25], and the strong bimetallic electronic ligand interaction between Au and Pd elements [20] also contributes to the superior catalytic performance over monometallic counterparts. The slower decay of steady-state current suggests that the electrode surface does not undergo deactivation during the electro-oxidation process. This indicates that the Au@Pd-G electrode has the best tolerance towards poisoning by CO-like intermediates. Therefore, the catalytic activity is in order of Au@Pd-G > Pd-G > Au-G ( Fig. 8(b)).
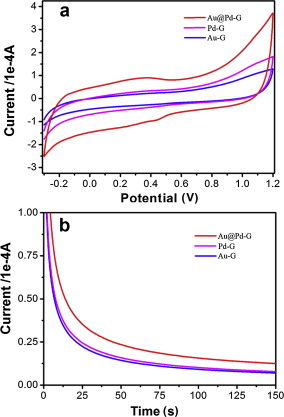 | Fig. 8 (a) CVs and (b) I-t curves of Au@Pd-G, Pd-G and Au-G modified GCEs in 100 mL mixed solution containing 0.5 mol/L H2SO4 and 0.5 mol/L N2-saturated CH3OH at a scan rate of 50 mV/s. |
In summary, graphene nanosheets supported bimetallic Au@Pd core-shell structured NPs can be synthetized by a simple one-step thermal reduction method. The bimetallic Au@Pd NPs with an average diameter of 11 nm can be homogeneously dispersed on the graphene surface. The Au core quantity as well as the Pd shell thickness can be quantitatively controlled by loading different amounts of metallic precursors, and the involved core-shell structure formation mechanism is also discussed. The ternary Pt/Au@Pd-G composites can also be synthetized by the subsequent Pt doping. The catalytic performance of Au@Pd-G composites toward methanol electro-oxidation in acidic media is investigated. The results show that Au@Pd-G composites exhibit higher catalytic activity, better stability and stronger tolerance to CO poisoning than Pd-G and Au-G counterparts. This method may be applied to synthetize graphene nanosheets supported other bimetallic NPs for applications in fuel cells.
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|