



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Thermal Annealing and Graphene Modification of Exfoliated Hydrogen Titanate Nanosheets for Enhanced Lithium-ion Intercalation Properties
[Xinning Luan, Ying Wang*  ]
]
]
|
|


Hydrogen titanate has been considered as a promising lithium intercalation material due to its unique layered structure. In the present work, we fabricate 2D graphene/hydrogen titanate hybrid nanosheets for application as anode materials in lithium-ion batteries. H2Ti3O7 nanosheets are synthesized by exfoliation of a layered precursor via interacting bulky tetrabutylammonium (TBA+) cations, followed by ion exchange with Na+ ions and washing with water. The as-prepared hydrogen titanate nanosheets are well-dispersed exhibiting ultra-thin thickness with a lateral size up to a few micrometers. The sample is then annealed at 450, 650 and 850 °C, to optimize its Li+-intercalation property. Heating at 450 °C leads to well-crystallized hydrogen titanate with a trace amount of TiO2. Heating at 650 and 850 °C results in mixed sodium titanates, since some sodium ions in the interlayer structure cannot be washed away and become chemically bonded to [TiO6] octahedra at high temperatures. Electrochemical properties of all the four samples are then evaluated by charged/discharged for 100 electrochemical cycles at 0.01-2.5 V
In the past decade, TiO2 and its derived nanomaterials have been intensively investigated owing to their multifunctional properties and wide applications in lithium-ion batteries[ 1], [ 2], [ 3] , [ 4], dye-sensitized solar cells[ 5], [ 6], [ 7], [ 8], [ 9] , [ 10], photocatalysis[ 11] , [ 12], and biosensors[ 13]. More recently, much attention has been paid to fabricating electrodes composed of alkali metal titanate family which is related to hydrogen titanate as well as their Li, Na, and K salts. Electrochemical behaviors of lithium intercalation into these titanate electrodes have been well addressed[ 14], [ 15], [ 16], [ 17], [ 18], [ 19], [ 20] , [ 21]. Titanates with layered structure are composed of corrugated chains of edge and corner sharing [TiO6] octahedra in which alkali ions occupy the interlayer spaces. This layered structure facilitates intercalation of Li+ ions by providing short and direct path for Li+ ions transport through interlayer spaces, and better accommodation of volume change caused by Li insertion/extraction. Therefore, titanates are considered as very promising active materials for applications in Li-ion batteries with high charge/discharge capacity, high-energy density, and long cycle life.
A common method to synthesize titanates is hydrothermal approach. Tsai and Teng obtained titanate nanotubes by using bulk TiO2 powder and NaOH as precursors in a hydrothermal process[ 22]. Titanate nanorods, nanowires, nanofibers, and nanobelts have also been prepared from TiO2 by using a simple hydrothermal treatment with alkaline followed by acid washing[ 23] , [ 24]. Among alkali metal titanates, hydrogen titanate with unique electrochemical performance is considered as a very promising lithium intercalation material for high-energy rechargeable Li-ion batteries and electrochemical supercapacitors[ 14], [ 15], [ 16], [ 25] , [ 26]. Li et al. reported a hydrothermal synthesis of layered hydrogen titanate nanowires with the composition of H2Ti3O7 and investigated their lithium intercalation properties for applications as electrodes in lithium-ion batteries[ 17]. The initial discharge capacity of the as-prepared micron-long H2Ti3O7 nanowires could reach 296.6 mA h g-1 at a specific current of 300 mA g-1, and could keep a very high capacity of 132.2 mA h g-1 even at a very high specific current of 2500 mA g-1. Such excellent electrochemical properties of hydrogen titanate are attributed to the open layered structure with a much larger interlayer spacing than common intercalation compounds. Wei et al. successfully synthesized layer-structured H2Ti3O7 nanowires with lengths up to several micrometers via hydrothermal process, which delivered a discharge capacity of 100 mA h g-1 at 40 A g-1 demonstrating excellent rate performance due to shorter Li-ion diffusion distance for their insertion/extraction into/from layered H2Ti3O7 nanowires[ 27]. Sodium titanates with a basic formula of Na2Ti nO2 n+1 have also been tested as anode materials for Li-ion batteries due to their good electrochemical lithium insertion properties. This material has a stable structure during Li-ion insertion/extraction processes with the presence of sodium in the pristine material[ 14]. Lithium can be reversibly inserted into rod-like Na2Ti6O13 with a capacity of 150 mA h g-1 at the C/3 rate[ 14]. For Na2Ti3O7, the initial Li-ion interaction capacity was only 44 mA h g-1 at a specific current of 10 mA g-1; this value was equivalent to 0.5 electron transfer per formula unit[ 15]. Rudola et al. recently evaluated electrochemical performance of Na2Ti3O7 for application as anode material in Na-ion batteries. Na2Ti3O7 electrode exhibited a first discharge capacity of 177 mA h g-1 and a relative low cycling stability with capacity retentions of 55.3% after 90 cycles at the 0.1 C rate over a voltage window of 0.01-2.5 V vs. Na/Na+ [ 28]. However, all the work summarized above either presented very low cycling stability of batteries based on alkali metal titanates or did not report any cycling performance. Moreover, among these reports, alkali metal titanates were obtained in the form of one-dimensional (1D) structure such as nanotube, nanowire, nanorod, and nanobelt. Even though 1D structure provides a direct pathway for Li-ion transport, these 1D nanostructures exhibited either defects or random alignments causing poor electronic transport during Li-ion insertion/extraction leading to low capacity and impeding its practical application.
Graphene, a single layer of two-dimensional carbon lattice, has recently emerged as a novel nanomaterial for applications in energy storage technology owing to its very light weight, large surface area, and extremely high electron mobility (~15,000 cm2 V-1 s-1) at room temperature[ 29], [ 30] , [ 31]. Such merits suggest that graphene nanosheets can be an ideal conductor to be mixed with metal oxides to improve their electrochemical performances. Graphene based nanocomposite electrode materials such as TiO2[ 32], [ 33] , [ 34], SnO2[ 35], [ 36] , [ 37], WO3[ 38], Co3O4[ 39] , [ 40], MnO2[ 41] etc. have been explored showing enhanced storage capacity and cycling stability. It has been demonstrated that incorporation of graphene into these composites greatly facilitates electrical conductivity by decreasing the charge transfer resistance of active material and prevents the aggregation of active material during cycling. However, there are few reports about nanocomposites based on graphene/alkali metal titanate nanosheets, due to the challenge of combining 1D nanostructure of alkali metal titanate with two-dimensional (2D) graphene nanosheets. One solution is to develop 2D titanate nanosheets which have similar morphology with graphene sheets and can thus hybridize with graphene more uniformly.
To the best of our knowledge, electrochemical property of hydrogen titanate with 2D nanosheet structure has never been studied. Herein, we fabricate 2D hydrogen titanate nanosheets via chemical exfoliation of a layered precursor through acid treatment and ion-exchange with bulky organic ions and Na+ ions, followed by washing with water. Thermal annealing of the as-prepared hydrogen titanate nanosheets at different temperatures is carried out to optimize their electrochemical properties for applications as anode materials in lithium-ion batteries. Moreover, graphene nanosheets are added to titanate nanosheets to further enhance their Li-ion intercalation properties owing to significantly increased electronic conductivity from graphene. Both titanate nanosheets and hybridization with graphene nanosheets are achieved for the first time. Electrochemical cycling performances of these materials are also explored, showing significantly enhanced Li-ion intercalation properties.
Lamellar solids of lepidocrocite-type cesium titanate Cs xTi2- x/4□ x/4O4 (□: vacancy, x = 0.7) was synthesized via a conventional solid-state calcination[ 42] , [ 43]. A stoichiometric mixture of Cs2CO3 (Alfa Aser, 99.99%) and TiO2 (anatase, 99%, Sigma Aldrich) was calcinated at 1073 K for 20 h with a molar ratio of 1:5.3. After cooling, the calcinated products were ground and calcinated repeatedly. Subsequent acid leaching converted them into protonated forms of H xTi2- x/4□ x/4O4·H2O[ 44] , [ 45]. The protonated titanate was derived through repeated ion exchange of Cs with proton. The resulted powder (~2 g) was stirred in 200 ml hydrochloric acid solution with a concentration of 1 mol l-1 for 24 h. After Cs extraction was completed via four cycles of ion exchange, the acid-treated product was thoroughly washed with water to remove acid residue and dried under ambient condition.
The as-prepared H xTi2- x/4□ x/4O4·H2O was treated with tetrabutylammonium hydroxide (TBAOH, (C4H9)4NOH, ~40% solution, Fluka) to delaminate into nanosheets. A weighed amount (2 g) of H xTi2- x/4□ x/4O4·H2O was shaken vigorously in an aqueous solution (500 ml) of TBA hydroxide, for two weeks at room temperature. The amount of TBA hydroxide was 5-fold excess to the exchangeable capacity of 4.12 meq g-1 for H xTi2- x/4□ x/4O4·H2O.
Typically, 100 ml colloidal suspension of TBA-intercalated titanate was poured into 100 ml of NaOH aqueous solution (1 mol l-1). Wool-like precipitates were yielded and the mixture was stirred overnight. After filtration and washing with distilled water, a post-calcination process is also necessary to remove organic residues and to form a high-crystalline phase. The obtained solids were then heated at 450, 650, and 850 °C to form alkali metal titanates. Then the resulted materials were shaken in 100 ml distilled water to remove excess sodium from interlayers of titanate host structure with washing using fresh water every 12 h for more than 3 times. Final products were collected from centrifugation, and then air-dried at room temperature. Hybrid graphene/hydrogen titanate nanosheets were fabricated by mixing graphene nanosheets (ACS Material, LLC) with hydrogen titanates in a weight ratio of 1:100.
Crystal structures of alkali metal titanates were determined by X-ray diffraction (XRD) using a Rigaku MiniFlex diffractometer with Cu Kα radiation operated at 30 kV and 15 mA with a scan rate of 2°/min. Morphology of titanates was characterized by scanning electron microscopy (SEM, FEI Quanta 3D FEG) at an accelerating voltage of 10 kV. Chemical composition of hydrogen titanate nanosheets annealed at three different temperatures was studied by energy dispersive X-ray spectroscopy (EDX) on an FEI Quanta 200 SEM. High-resolution transmission electron microscopic (HRTEM) images of the as-prepared titanates were taken using JEOL HRTEM (JEM-1400 electron microscope) with an acceleration voltage of 120 kV. The samples were also observed under a polarized optical microscope using an Olympus BX-51 microscope.
For electrochemical measurements, the as-prepared titanates were mixed with acetylene black (AB, Alfa Aesar, 99.5%) and polyvinylidene fluoride (PVDF, Alfa Aesar) at a weight ratio of 70:20:10 in N-methyl-2-pyrrolidene (NMP) solvent. Graphene-modified titanates were synthesized by mixing graphene nanosheets (ACS Material) with titanates with a weight ratio of 1:100. The resultant viscous slurry was coated on a copper foil using an AFA-III automatic film applicator (MTI Corporation) with a thickness setting of 500 μm and then dried at 120 °C overnight under vacuum, followed by pressing and cutting into a disc shape. The electrodes were assembled into CR2032-type coin cells in an argon filled glovebox using lithium foil as the counter electrode and a microporous membrane (Celgard-2320, USA) as the separator; electrolyte was 1 mol l-1 lithium hexafluorophosphate (LiPF6) dissolved in a mixed solvent of ethylene carbonate, dimethyl carbonate, and diethyl carbonate (1:1:1 volume ratio). The coin cells were galvanostatically charged and discharged between 0.01 and 2.5 V vs. Li+/Li at a specific current of 170 mA g-1 using an 8-channel battery analyzer (MTI Corporation).
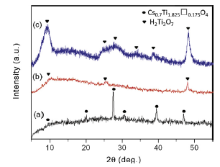
Fig. 1 shows XRD patterns of the materials before and after exfoliation: (a) calcinated cesium titanate, (b) TBA+-intercalated titanate (dried sample), and (c) exfoliated product obtained after ion exchange with NaOH and washing with water. XRD pattern in Fig. 1(a) shows that the calcinated product is identified as a homogeneous single phase of lepidocrocite-type cesium titanate Cs0.7Ti1.825□0.175O4 (JCPDS #40-0827) which is synthesized using TiO2 and Cs2CO3 with a molar ratio of 5.3:1 followed by heat treatment at 800 °C for 20 h. As reported by Sasaki et al.[ 44] , [ 45], the protonated titanate H xTi2- x/4□ x/4O4·H2O is then produced through repeated ion exchange of Cs ions with protons. Afterwards, acid-treated H xTi2- x/4□ x/4O4·H2O reacts with TBAOH, intercalating TBA+ ions into the interlayer space of H xTi2- x/4□ x/4O4·H2O through ion exchange of TBA with protons. The TBA+-intercalated titanate is dried at room temperature and shows relatively small XRD diffraction peaks in Fig. 1(b); this crystalline phase is probably an intermediate phase of final product. Fig. 1(c) displays XRD pattern of exfoliated product obtained after ion exchange of TBA+ with Na+ ions and washing with water. The diffraction peaks of exfoliated product cannot be indexed to any known standard JCPDS pattern, but similar to those of hydrogen titanate with the composition of H2Ti3O7 reported in literatures[ 11] , [ 17]. Hydrogen titanate crystallizes in a monoclinic system with a space group ( C2/m) and a layered structure. As can be seen in Fig. 1, XRD patterns of TBA+-intercalated titanate ( Fig. 1(b)) and H2Ti3O7 ( Fig. 1(c)) are quite similar, suggesting that the skeleton structure is retained upon ion exchange with TBA+ ions. Compared with XRD pattern of TBA+-intercalated titanate, the diffraction peaks of hydrogen titanate are much sharper and stronger indicating better crystallization after Na+ substituting TBA+ ions and washing with water.
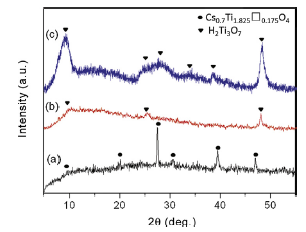 | Fig. 1. Fig. 1. X-ray diffraction patterns of samples before and after exfoliation: (a) calcinated cesium titanate, (b) TBA+-intercalated titanate (dried sample), and (c) exfoliated hydrogen titanate obtained via TBA+ intercalation into protonated titanate and ion exchange with NaOH followed by washing with water. |
Morphologies of cesium titanate and exfoliated hydrogen titanate are investigated via SEM. As can be seen from Fig. 2(a), cesium titanate consists of plate-like particles with widths at sub-micron scale and lengths up to one micron. Others have reported that protonated titanate is stabilized in a suspension containing TBA+OH-; in this system, H+ ions in the interlay structure of titanate are replaced by much larger TBA+ ions, increasing the interlayer space of titanate[ 44] , [ 45]. During the flocculation using NaOH solution, Na+ ions substitute TBA+ ions. The nanosheet colloidal suspension becomes unstable and the nanosheets become stacked together by intercalating Na+ ions into the interlayer space. When shaking in water, Na+ ions in the interlayer are gradually deintercalated. During vigorous stirring, the interlayer spacing might also be expanded due to intercalation of water and the increased water content in the interlayer space, and thus significantly reducing the electrostatic interaction between neighboring sheets[ 46]. As can be seen from Fig. 2(b), hydrogen titanate is delaminated and the resulted sheets are well dispersed. Most of the dispersed nanosheets exhibit lateral curling indicating successful delamination of hydrogen titanate.
 | Fig. 2. Fig. 2. SEM images of (a) calcinated cesium titanate and (b) exfoliated hydrogen titanate obtained via TBA+ intercalation into protonated titanate and ion exchange with NaOH followed by washing with water. |
TEM images in Fig. 3(a) and (b) further reveal dimensions and structural details of titanate nanosheets. Fig. 3(a) shows TEM image of hydrogen titanate nanosheets with a lateral size up to micron scale, which is consistent with the size of the starting material (cesium titanate) that is used to synthesize hydrogen titanate. Fig. 3(a) and (b) both display hydrogen titanate nanosheets that are translucent or almost transparent, indicating their ultra-thin thickness. The inset image in Fig. 3(b) shows the layered structure of hydrogen titanate with an interlayer spacing of 0.8 nm, which is consistent with what has been reported in literature[ 17]. Additionally, crystallinity of TBA+-intercalated titanate suspension is detected by eyes and polarized microscopy due to birefringence of colloids. Fig. 3(c) shows a photograph of TBA+-intercalated titanate colloidal dispersion, which appears translucent and homogeneous. This colloidal dispersion is stable for weeks, indicating the nanosheets with ultra-thin thickness are uniformly dispersed and stable in the solvent. Fig. 3(d) shows a polarized microscopy image of TBA+-intercalated titanate nanosheet colloidal dispersion, in which liquid crystallinity of TBA+-intercalated titanate nanosheet suspension is confirmed by birefringence of colloids. However, crystallinity observed by polarized microscopy is not uniform, indicating that the TBA+-intercalated titanate nanosheets may not be completely crystallized. Therefore, a post-calcination process is needed to form a well-crystalline phase.
 | Fig. 3. (a, b) High-resolution TEM images of exfoliated hydrogen titanate, the inset showing layered structure of hydrogen titanate; (c) photograph and (d) polarized microscopic image of TBA+-intercalated titanate colloidal dispersion. |
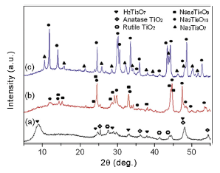
After intercalation with TBA+, ion exchange with Na+, washing with water and exfoliation, the resulted hydrogen titanate is subjected to heat treatment in order to remove organic residues such as TBA+ ions and to form a well-crystalline phase. Fig. 4 presents XRD patterns of hydrogen titanate annealed at different temperatures (450, 650, and 850 °C) for 3 h, indicating that different compositions are obtained during annealing at different temperatures. XRD pattern of the sample annealed at 450 °C for 3 h can be indexed to original phase of H2Ti3O7 with trace impurities of rutile TiO2 and anatase TiO2. Fig. S1 displays EDX spectra of hydrogen titanate nanosheets annealed at different temperatures (450, 650, and 850 °C). EDX spectra from all the three samples show main peaks of Ti and O and low peaks of Na and C. Presence of carbon is attributed to carbon tape used on the sample holder, leaving Ti, O, Na from the samples. Results from EDX and XRD characterizations indicate that a small amount of Na+ ions is intercalated in the interlayer space of hydrogen titanate and remain there even after being annealed at 450 °C. It is because Na+ ions cannot be completely extracted by washing the titanate with water after ion exchange with Na+ ions. Thus, the product after ion exchange is described as Na+ intercalated H2Ti3O7 or Na xH2- xTi3O7[ 47] , [ 48]. Even though the intercalated Na ions do not change the layered structure of hydrogen titanate after being annealed at 450 °C as shown in Fig. 4(a), XRD peaks detected in Fig. 4(b) and (c) suggest formation of sodium titanate compounds after the sample is annealed at higher temperatures of 650 and 850 °C. When the calcination temperature is increased to 650 °C, XRD diffraction pattern of the sample matches mixture of Na2Ti6O13 (JCPDS #37-0951) and Na0.8Ti4O8 (JCPDS #73-1400) with monoclinic phase. Formation of these compounds can be explained as follows. Annealing at 650 °C could cause Na+ ions chemically bonded to [TiO6] octahedral layers, yielding sodium titanate. XRD pattern of the sample annealed at 850 °C seems to be similar to that of Na2Ti6O13 but also shows impurities of monoclinic Na2Ti3O7 (JCPDS #72-0148). Na0.8Ti4O8 is present at 650 °C but disappears after annealing at 850 °C, possibly because it is an intermediate phase. It is also noted that hydrogen titanate and sodium titanates have a common monoclinic structure, with a crystal lattice consisting of [TiO6] octahedra sharing edges and zigzag chain-like structure[ 13], [ 15] , [ 49]. These chains are joined together by sharing edges to form layers, while protons or Na+ ions can reside between the layers. With increasing annealing temperature, more intercalated Na+ ions chemically bond to [TiO6] octahedral structure in the titanate, resulting in sodium titanate with higher ratio of Na to Ti.
 | Fig. 4. XRD patterns of hydrogen titanate annealed at different temperatures for 3 h: (a) 450 °C (H2Ti3O7 with trace amounts of rutile and anatase TiO2), (b) 650 °C (Na2Ti6O13 mixed with Na0.8Ti4O8), and (c) 850 °C (Na2Ti6O13 mixed with Na2Ti3O7). |
Morphologies of annealed hydrogen titanate at different temperatures (450, 650, and 850 °C) are shown in Fig. 5. At 450 °C, the delaminated hydrogen titanates are restacked together. At 650 °C, due to bonding between Na+ ions and [TiO6] octahedral layers, the original anisotropic structure induces formation of rod-like sodium titanate particles with widths at sub-micron scale and lengths at micron-scale. Annealing at an even higher temperature of 850 °C leads to agglomeration of micro-sized sodium titanate particles.
 | Fig. 5. SEM images of hydrogen titanate annealed under different temperatures (a) 450 °C (H2Ti3O7 with trace amounts of rutile and anatase TiO2), (b) 650 °C (Na2Ti6O13 mixed with Na0.8Ti4O8), and (c) 850 °C (Na2Ti6O13 mixed with Na2Ti3O7). |
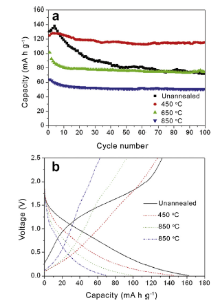
Electrochemical performances of the as-synthesized hydrogen/sodium titanates are evaluated by charged/discharged in a voltage range of 0.01-2.5 V vs. Li/Li+ at a specific current of 170 mA g-1 for 100 cycles. Fig. 6(a) and (b) compares respectively cycling performances and the corresponding 2nd charge and discharge curves of unannealed hydrogen titanate and hydrogen titanate annealed at different temperatures (450, 650, and 850 °C). The unannealed hydrogen titanate delivers the highest initial discharge capacity of 130.5 mA h g-1, which is higher than 124.6 mA h g-1 from hydrogen titanate annealed at 450 °C, as well as 101.3 and 63.8 mA h g-1 from hydrogen titanate annealed at 650 and 850 °C, respectively. As shown in Fig. 6(b), the 2nd charge and discharge capacities from unannealed hydrogen titanate are quite large, which can be ascribed to bonded water in the layered unannealed hydrogen titanate. The increased interlayer space resulted from water between layers in the layered structure allows insertion/extraction of more Li ions into/from unannealed hydrogen titanate, leading to high specific charge and discharge capacities. Also, high surface area from the well-dispersed nanosheets provides a shorter Li-ion diffusion length, more direct pathway for electron transport, and larger electrode/electrolyte contact area of well-dispersed nanosheets compared to micro-sized rods or agglomerates in other annealed samples, which facilitates Li-ion insertion and extraction. However, during subsequent electrochemical cycles, unannealed hydrogen titanate suffers from faster capacity fading. In contrast, hydrogen titanate annealed at 450 °C retains a charge capacity of 115.2 mA h g-1 after 100 cycles, corresponding to a capacity retention of 92.5%, while unannealed hydrogen titanate exhibits a final capacity of 72.1 mA h g-1 and a very poor capacity retention of 55.2%. Such faster capacity fading of unannealed hydrogen titanate can be ascribed to imperfect crystalline phase synthesized via exfoliation process ( Fig. 1(c)). After heat treatment, organic residues such as TBA+ ions would be removed, which enhances the cycling stability. Micro-sized sodium titanates synthesized by annealing hydrogen titanate at 650 and 850 °C exhibit mediocre cycling stability with capacity retentions of 73.8% and 79.1%, respectively, due to less surface area from large particles or agglomerates. In addition, different chemical compositions of the three samples can affect specific capacities due to their different theoretical Li-ion interaction capacities. Because hydrogen titanate is lighter than sodium titanate in molar weight, the former holds larger reversible capacity of 200 mA h g- 1 [ 17]. Also, lithium insertion into Na2Ti6O13 is facilely reversible with a maximum 3 mol of lithium intercalated per formula unit, corresponding to a capacity of 150 mA h g- 1 [ 14]. However, Na2Ti3O7 has a low capacity of 44 mA h g-1 corresponding to 0.5 mol per formula unit[ 15]. Therefore, when the annealing temperature is increased to 850 °C, the formation of Na2Ti3O7 leads to capacity decrease.
 | Fig. 6. (a) Cycling behaviors and (b) 2nd charge and discharge curves of unannealed hydrogen titanate and hydrogen titanate annealed at different temperatures (450, 650, and 850 °C), cycled in a voltage range of 0.01-2.5 V vs. Li/Li+ at a specific current of 170 mA g-1 for 100 electrochemical cycles. |
To further improve electrochemical performance, graphene/hydrogen titanate hybrid nanosheets are fabricated by mixing graphene nanosheets with hydrogen titanates in a weight ratio of 1:100. Fig. 7(a) shows SEM image of unannealed hydrogen titanate nanosheets hybridized with graphene sheets, which looks like stickers (titanate sheets) attached on larger pieces of wrinkled paper (graphene sheets). Fig. 7(b) displays SEM image of hydrogen titanate annealed at 450 °C hybridized with graphene, showing graphene nanosheets covered by densely packed hydrogen titanates, since heat treatment at 450 °C causes hydrogen titanate nanosheets to stack up and agglomerate.
 | Fig. 7. SEM images of (a) unannealed hydrogen titanate hybridized with graphene, (b) hydrogen titanate annealed at 450 °C and hybridized with grapheme. |
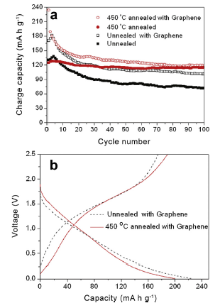
Fig. 8(a and b) compares respectively cycling performances and the corresponding 2nd charge and discharge curves of unannealed hydrogen titanate and hydrogen titanate at 450 °C with and without graphene modification in a voltage range of 0.01-2.5 V vs. Li/Li+ at a specific current of 170 mA g-1. It is noted that graphene-modified hydrogen titanates (unannealed and annealed at 450 °C) show enhanced capacities than those of unmodified hydrogen titanates. It is observed that unannealed hydrogen titanate modified with graphene delivers an initial charge capacity of 170.7 mA h g-1 and retains a charge capacity of 101.0 mA h g-1 after 100 electrochemical cycles. Compared to the final capacity (72.1 mA h g-1) from unmodified hydrogen titanate, an enhanced charge capacity of 28.9 mA h g-1 is achieved with graphene modification, representing an enhancement of 40%, due to remarkably increased electronic conductivity from graphene and reduced charge transfer resistance in the hybrid. Moreover, the well-dispersed hydrogen titanate nanosheets can combine with graphene nanosheets uniformly due to their similar 2D structure and thus retard aggregation of active material. It can also be seen from Fig. 8 that hydrogen titanate annealed at 450 °C and modified with graphene delivers an initial charge capacity of 233.9 mA h g-1 but a much lower final capacity of 119.7 mA h g-1 after 100 electrochemical cycles. Compared to hydrogen titanate annealed at 450 °C without graphene modification exhibiting an initial capacity of 124.6 mA h g-1, initial capacity is greatly increased by 187% due to graphene modification, but faster capacity fading is also observed after graphene modification. The reason for less distinct enhancement in retained capacity after 100 cycles can be attributed to agglomeration of hydrogen titanates caused by heat treatment at 450 °C.
 | Fig. 8. (a) Cycling behaviors and (b) 2nd charge and discharge curves of unannealed hydrogen titanate and hydrogen titanate annealed at 450 °C with and without graphene modification, cycled in a voltage range of 0.01-2.5 V vs. Li/Li+ at a specific current of 170 mA g-1 for 100 electrochemical cycles. |
In summary, hydrogen titanate nanosheets are synthesized by ion exchange between hydrogen acid with cesium titanate followed by exfoliation via interclalation of TBA+ ions and ion exchange with Na+ and washing with water, for applications as anode materials in lithium-ion batteries. Subsequent heat treatment at 450 °C transforms them to well-crystalline phase for enhanced electrochemical property, though heat treatment at higher temperatures (650 and 850 °C) results in lower capacities. Hybridization with graphene nanosheets leads to further enhanced electrochemical performance, due to increased electronic conductivity from graphene. Composition and morphology of unannealed hydrogen titanate and hydrogen titanate annealed at different temperatures (450, 650, and 850 °C) are investigated by XRD, SEM, and TEM. It is found that heat treatment at 450 °C removes organic residues, yielding well-crystalline phases H2Ti3O7 with trace amounts of rutile TiO2 and anatase TiO2. Since some Na+ ions are still intercalated in the interlayer space of the as-prepared hydrogen titanate nanosheets, heat treatment at higher temperatures (650 and 850 °C) causes Na chemically bonded to [TiO6] octahedral layer, producing monoclinic sodium titanates (Na2Ti6O13 mixed with Na0.8Ti4O8 at 650 °C, Na2Ti6O13 mixed Na2Ti3O7 at 850 °C). The four samples are then evaluated as Li-ion battery anode materials using electrochemical measurements. The unannealed hydrogen titanate delivers the highest initial discharge capacity but the poorest cycling performance. The sample annealed at 450 °C shows the highest capacity retention and the highest final capacity after 100 electrochemical cycles owing to removal of organic residues at this temperature, while samples annealed at 650 and 850 °C show very low capacities and mediocre cycling performances due to formation of sodium titanates and large particle size or agglomeration of particles at higher temperatures. To further improve electrochemical performance of hydrogen titanate, graphene/hydrogen titanate hybrid nanosheets are synthesized by mixing graphene with the unannealed hydrogen titanate and hydrogen titanate annealed at 450 °C. The initial charge capacities of both hybrids are largely increased due to significantly improved electronic conductivity. Modified by graphene nanosheets, unannealed hydrogen titanate exhibits more distinct enhancement in capacity due to uniform combination of well-dispersed hydrogen titanate nanosheets with graphene nanosheets resulted from their similar 2D structure. Our results confirm that exfoliated hydrogen titanate nanosheets after thermal annealing and graphene modification can be promising anode materials for applications in lithium-ion batteries.
APPENDIX A. SUPPLEMENTARY DATA
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.jmst.2014.07.003.
Acknowledgments
This work is supported by LABOR-RCS grant. The authors acknowledge Materials Characterization Center at LSU for using XRD and SEM. X.N. Luan also acknowledges LSU Graduate School Enrichment Award.
| 1. |
|
| 2. |
|
| 3. |
|
| 4. |
|
| 5. |
|
| 6. |
|
| 7. |
|
| 8. |
|
| 9. |
|
| 10. |
|
| 11. |
|
| 12. |
|
| 13. |
|
| 14. |
|
| 15. |
|
| 16. |
|
| 17. |
|
| 18. |
|
| 19. |
|
| 20. |
|
| 21. |
|
| 22. |
|
| 23. |
|
| 24. |
|
| 25. |
|
| 26. |
|
| 27. |
|
| 28. |
|
| 29. |
|
| 30. |
|
| 31. |
|
| 32. |
|
| 33. |
|
| 34. |
|
| 35. |
|
| 36. |
|
| 37. |
|
| 38. |
|
| 39. |
|
| 40. |
|
| 41. |
|
| 42. |
|
| 43. |
|
| 44. |
|
| 45. |
|
| 46. |
|
| 47. |
|
| 48. |
|
| 49. |
|