{kind=link}
{kind=link}
{kind=link}
Theoretical Investigation on Mechanical and Thermal Properties of a Promising Thermal Barrier Material: Yb3Al5O12
[Zhou Yanchun*  , Xiang Huimin, Feng Zhihai]
, Xiang Huimin, Feng Zhihai]
, Xiang Huimin, Feng Zhihai]
|
|


An investigation on the mechanical and thermal properties of Yb3Al5O12 is conducted by a combination of first-principles calculations and chemical bond theory calculation. Density functional theory (DFT) computations were performed for the structural, mechanical, and thermal properties, and the results are confirmed by chemical bond theory. Based on the calculated equilibrium crystal structure, heterogeneous bonding nature is revealed. The full set of elastic constants and mechanical properties of Yb3Al5O12 are presented for the first time. The thermal expansion coefficient of Yb3Al5O12 is calculated to be 7.5 × 10-6 K–1 by chemical bond theory. In addition, the minimum thermal conductivity of Yb3Al5O12 is estimated to be 1.22 W m-1 K-1, and the origin of such low thermal conductivity is discussed. Our theoretical results highlight the potential of Yb3Al5O12 as a prospective thermal barrier coating material.
The efficiency and power of gas-turbine engines used in transportation, energy, and defense sectors rely on the increase of the operated temperature [1]. To increase the operating temperatures of engines, efficient thermal barrier coatings (TBCs) are needed [2]. The current material of choice for TBCs is yttria-stabilized zirconia (YSZ) in its metastable tetragonal-prime structure [3]. However, the working temperature of YSZ is limited because the metastable tetragonal-prime structure becomes increasingly unstable at high temperature and is apt to decompose to a mixture of tetragonal and cubic zirconia [4]. Additionally, the oxygen diffusivity of YSZ is high at high temperature [5]. Therefore, new TBCs with superior high temperature properties are needed for both scientific significance and technological importance.
Experimental and theoretical trials reported recently have proved that yttrium aluminates, and yttrium aluminum garnet (i.e. Y3Al5O12, YAG) in particular, are promising candidates for TBCs due to their superior high temperature stability, mechanical and thermal properties [6] and [7]. At high temperature, YAG is stable with Al2O3 [6] that is the thermally grown oxide formed on Ni-based superalloys. High-temperature deformation tests on single-crystal YAG showed that YAG is one of the most creep-resistant, non-oxidizing material yet known [8] and [9]. Recent theoretical results have confirmed the low thermal conductivity of YAG [10]. Thus, YAG is considered as a promising TBC material. However, Klemm et al. [11]found that the resistance of YAG to high temperature water corrosion is not quite satisfactory, which is adverse on the integrity and duration of coating. Therefore, materials with improved resistance to vapor corrosion are needed.
In the same work, Klemm et al. [11] reported that Yb3Al5O12(YbAG), which shares the same structure with YAG, have superior resistance to vapor corrosion comparing with several other prospective TBC materials, such as Y3Al5O12, Y2Si2O7and Yb2Si2O7. Having the same structure with YAG, YbAG is expected to possess similar excellent high temperature properties and lower minimum thermal conductivity due to the heavy atomic mass of Yb. Motivated by its potential high temperature applications, several works focused on the preparation of YbAG have been reported recently, by various approaches, such as co-precipitation method [12], micro-pulling-down technique [13], and RF-heating Czochralski method [14]. However, to the present authors' knowledge, the mechanical and thermal properties of YbAG have barely been reported. Clear understanding of mechanical and thermal properties and correlation of these properties to the structure of YbAG can provide valuable insights for the development of TBCs, and advance the applications of YbAG as high temperature structural component and TBC.
In this contribution, mechanical and thermal properties of Yb3Al5O12were investigated from a combination of first-principles calculations and an empirical method based on chemical bond theory. The equilibrium crystal structure, second-order elastic coefficients, bulk modulus, shear modulus, Young's modulus, and Poisson's ratio were calculated by first-principles calculations. Then, the theoretical minimum thermal conductivity was estimated from modified Clarke's model [15] and [16], and the dependence of thermal conductivity of Yb3Al5O12on temperature was predicted from Slack's model [17]. To valid the theoretical calculation, bulk modulus of Yb3Al5O12 was estimated using chemical bond theory. The average linear thermal expansion coefficient (TEC) was also obtained using this empirical method. In the end, we highlight the potential application of Yb3Al5O12as TBC.
The first-principles calculations based on the density functional theory (DFT) were performed using the Cambridge Serial Total Energy Package (CASTEP) code [18]. Wherein the Norm-conserving pseudopotentials [19] were employed to represent the interactions between the ionic cores and the valence electrons for Yb, Al and O atoms. The electronic exchange-correlation energy was treated under the local density approximation (LDA) [20]. Feng et al. [21] investigated the effects of onsite Coulomb interaction ( U correction) on the electronic structures and mechanical properties of Re2Zr2O7 pyrochlore. They found that the additional U correction only strongly affects the electronic structures of the rare-earth oxides, while the mechanical moduli are quite insensitive to the localization of partially filled 4f orbitals. Thus, the theoretical investigations in our work were conducted without consideration of U correction. The plane-wave basis set cut-off energy was fixed at 830 eV in all calculations after convergence test. The special points sampling integration over Brillouin zone are realized by using the Monkhorst–Pack method with special k-points meshes of 2 × 2 × 2 [22].
To get the equilibrium crystal structure, the lattice parameters and internal atomic coordinates were independently modified. The free enthalpy, interatomic forces and stresses of the unit cell were minimized under the Broyden–Fletcher–Goldfarb–Shanno (BFGS) minimization scheme [23]. The tolerances for geometrical optimization were: differences for total energy within 5 × 10-6eV/atom, maximum ionic Hellmann–Feynman force within 0.1 eV/nm, maximum ionic displacement within 5 × 10-5nm, and maximum stresswithin 0.02 GPa.
The theoretical elastic coefficients were determined from the first-principles strain–stress relationships method implemented by Milman et al. [24]. The criteria for convergence of optimization on atomic internal freedoms were selected as differences in the total energy within 1 × 10-6eV/atom, Hellmann–Feynman force within 0.02 eV/nm, and maximum ionic displacement within 1 × 10-5nm. The compliance tensor S was calculated as the inverse of the stiffness matrix, S = C-1. The bulk modulus B and shear modulus G were calculated from the compliance tensor based on Voigt–Reuss–Hill approximation [25], [26] and [27]. The Young's modulus EH and Poisson's ratio ν were calculated using Hill's bulk modulus BHand shear modulus GH by the following equations [28]:
According to Slack's model [17] and modified Clarke's model [15] and [16], the behavior of thermal conductivity at elevated temperatures of a material is accessible. Slack's model describes the intrinsic thermal conductivity of a material over wide range of temperatures, which is expressed as [17]:
where A is a physical constant ( A ≈ 3.1 × 10-6 if κL is in W m-1K-1, and δ in nm).
According to the modified Clarke's model [15] and [16], the theoretical lower limit of intrinsic thermal conductivity ( κmin) can be calculated by:
where kB is the Boltzmann's constant, υmthe average sound velocity, NAV the Avogadro's number, d the density, M the molecular weight, and n the number of atoms in the molecule. This modified model has been successfully applied to estimate the κmin of several complex oxides, such as La2T2O7 [16], M P2O7 [30], and MPO4 [31].
The average sound velocity υ m of a polycrystalline material is defined as [32]:
where υl and υs are longitudinal and transverse sound velocities, respectively, which can be determined from the shear modulus G and the bulk modulus B by:
The Debye temperature, ΘD, was calculated according to the following equation [32]:
where h is the Plank's constant.
Developed by Phillips [33], Van Vechten [34], Levine [35], and Zhang et al. [36], complex chemical bond theory (CBT) provides a simple but efficient approach to study the characteristic of chemical bonds in complex materials, and to predict related physical properties of a crystal from the viewpoint of bonding. The implementation of this theory relies on the decomposition of complex crystal into a linear combination of subformula of various binary crystals [36].
For a giving chemical formula of a complex crystal, CBT provides an efficient way to decompose it into the following simple subformula [36]:
where NC( A i) and NC( B j) represent the coordination numbers of A iand B jions in the crystal. N ( B j- A i) is the nearest coordination fraction contributed by A iion and vice versa. ( A m B n) μis the μ type of subformulas.
For every fictitious binary crystal ( A m B n) μdecomposed according to CBT, the total lattice energy Uμ can be separated into the ionic part
where Z + and Z - are the valence of cation and anion in the binary crystal, lμ is the length of μ type bond.
where
where bμ is a structural correction factor, rμ = lμ /2 is the average ion radius expressed in angstroms,
The lattice energy density uμ of a binary crystal, which is an essential parameter in estimating the bulk modulus and linear thermal expansion coefficient of a binary crystal, is defined as [38]:
where nμ is the number of the chemical bond in oneformula unit,
where δμ is a constant, and βμ is a proportion factor depending on the average valence and average coordination number of subformula A m B n. The bulk modulus of the complex crystals is expressed as [39]:
where B and Bμ are the bulk modulus of the material and μ type chemical bond, respectively, and V m and Vμ are the volume of one molecule and μ type bond in one molecule, respectively.
The linear TEC of a complex crystal is closely related to the lattice energy and can be estimated by the following equations [40]:
where parameter Δ μ is a correction parameter from the analysis of experimental results, which depends on the position of cation in the periodic table. Fμ is the fraction of μ type bond in the complex crystal.
As shown in Fig. 1, the crystal structure of Yb3Al5O12 which crystallizes in Ia
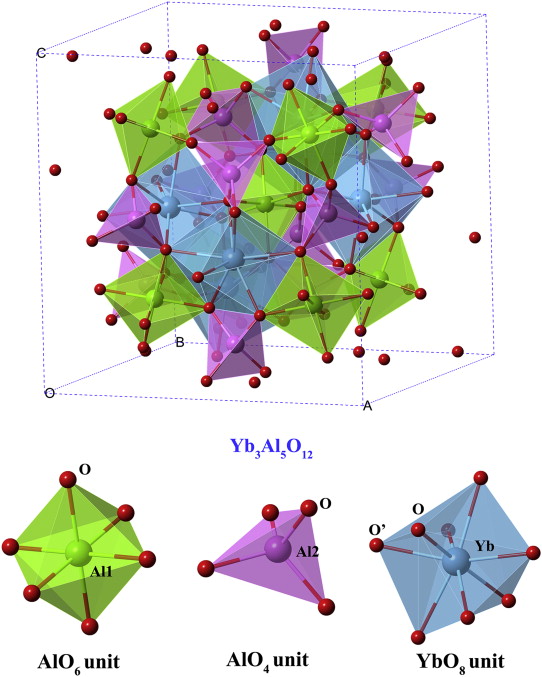 | Fig. 1. Crystal structure of Yb3Al5O12 and typical constructive units in Yb3Al5O12. |
| Table 1. Theoretical and experimental lattice parameters of Yb3Al5O12 |
The calculated lattice parameters of Yb3Al5O12 are listed in Table 1, together with previously reported experimental data [41] for comparison. As shown in Table 1, the calculated lattice parameter a is about 0.28% smaller than the experimental data. The atomic position of O atom is quite consistent with the reported one. Good coincidence between the theoretical results and the experimental ones ensures the reliability of our calculations.
The Mulliken analysis on Yb–O and Al–O bonds of Yb3Al5O12is also conducted by DFT calculations, and listed in Table 2. The bond lengths and Mulliken population of Yb–O and Al–O bonds of another rare-earth aluminate with the same structure, Y3Al5O12, is also included for comparison [10]. For both aluminates, the Mulliken population of Al–O bonds are higher than that of Yb/Y–O bonds, indicating higher level of covalency of Al–O bonds. Moreover, two types of Al–O bonds are quite different in bond length and Mulliken population, demonstrating that bond strength and covalency of Al–O in different polyhedra are quite different from each other.
| Table 2. Mulliken population and bond lengths of different bonds in Y3Al5O12 and Yb3Al5O12 |
For Yb3Al5O12, the chemical bonds are diverse in length and covalency, the bond strength and bond energy are anticipated to be miscellaneous. To quantify this diversity, the chemical parameters, such as covalency and bond energy, of each bond are calculated using CBT. First, based on the analysis on the crystal structure, the chemical formula of Yb3Al5O12is decomposed into the following subformulas:
Yb3Al5O12=YbAl(1)Al(2)O=Yb3/2O3+Yb3/2O3′ +Al(1)2O3+Al(2)3O3 (24)
where the superscripts in the formula mark the oxygen atoms having the same crystallography site with different bond lengths in one polyhedron. Then, the desired parameters of subformula are calculated by Eqs. (11), (12), (13), (14), (15), and) and are listed in Table 3. The bond lengths ( lμ) of all bonds used in the calculation of bond parameters are from the previously reported experimental data [41]. For all these bonds, the heteropolar part of energies ( Cμ) are substantially larger than the homopolar part of energies
| Table 3. Chemical parameters of subformulas for Yb3Al5O12 by chemical bond theory |
In general, the bonding nature is diverse and heterogeneous in Yb3Al5O12 though it crystallizes in cubic structure. Al–O units are strongly constructed and show rigid characteristic while YbO8 dodecahedron is relatively weak and soft. These structural features play an essential role in the elastic and thermal properties of this material, which will be discussed in the following sections.
To disclose the mechanical properties of Yb3Al5O12, which are closely related to the characteristic of crystal structure, the full sets of independent second-order elastic constants were calculated first. To the author's knowledge, the full sets of elastic constants of ytterbium aluminate have not been reported experimentally or theoretically so far. The calculated elastic constants are listed in Table 4, accompanied by those of Y3Al5O12as comparison. The mechanical stability of a crystal structure can be justified from the Born–Huang criterion [43]. For a cubic structure, the conditions could be expressed as: c44>0, c11>| c12|, and c11+ 2 c12>0. The criterions are completely satisfied by Yb3Al5O12, demonstrating its stability under elastic strain perturbations. Among all constants, c11represents the stiffness against principal strains while c44corresponds to resistance to shear deformation. A common feature for both materials is that c11is much higher than c44, indicating less resistance of these two compounds to the shear deformation.
| Table 4. Theoretical second-order elastic constants cij (in GPa), and Zener ratio ( A) of Yb3Al5O12 ceramic. The experimental data for single-crystal Y3Al5O12 is also included for comparison |
The calculated elastic constants are used to estimate the mechanical properties, i.e. bulk and shear moduli, Poisson's ratio and Young's modulus, according to Voigt–Reuss–Hill approximation [25], [26] and [27]. The calculated mechanical properties are tabulated in Table 5. However, no experimental data are available for Yb3Al5O12. Therefore, to verify our first principle calculation, the bulk modulus of Yb3Al5O12is also estimated using CBT. The calculated results are listed in Table 6. It is obvious that the bulk moduli for Al–O bonds are enormously larger than those of Yb–O bonds, showing less compressibility of Al–O bonds. The bulk modulus for polycrystal is estimated to be 164 GPa, which shows an excellent agreementwith the value (163 GPa) calculated by first-principles.
| Table 5. Theoretical bulk modulus BH (GPa), shear modulus GH (GPa), Young's modulus E (GPa), Poisson's ratio ( ν) and Pugh's ratio ( G/B) of Yb3Al5O12 ceramic |
| Table 6. Estimation of bond volume νμ (10-3 nm3), lattice energy density uμ (GPa), bulk modulus B (GPa) and linear thermal expansion coefficient α (×10-6 K–1) of Yb3Al5O12 from chemical bond theory |
Shear modulus G describes the resistance of a material against a shape change. For both materials, the shear moduli are quite smaller than the bulk modulus, indicating low resistance to shear deformation, as shown in Table 5. The G/B ratio (namely Pugh's ratio) is 0.64 and 0.60 for Yb3Al5O12 and Y3Al5O12, respectively. It is widely accepted that the ductility of a material could be reflected by Pugh's ratio. A low (high) Pugh'sratio value is associated with ductility (brittleness). The Pugh's ratios of SiC, a brittle ceramic, are 0.85 [44], and 0.76 for ZrC [45], while for a typical ductile ceramic, Ti3SiC2, it is 0.65 [46]. The Pugh's ratios of Yb3Al5O12and Y3Al5O12 are close to that of Ti3SiC2, indicating the possible damage tolerance for their low shear moduli.
The intrinsic hardness of a material is a highly complex property, which is difficult to describe with a formal theoretical definition. Chen et al. [47]proposed an empirical model, which is correlated not only with shear modulus G , but also with bulk modulus B. In their model, the Vickers hardness ( HV) is estimated by the following formula:
where k is the Pugh's modulus ratio. They found that their model performed well across a large class of materials and hardness values [47]. Based on chemical bond theory, Gao et al. [48]proposed another empirical equation to estimate hardness of covalent crystals: |
where
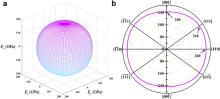
Young's modulus of polycrystalline Yb3Al5O12is 257 GPa according to Eq. (1). Based on the structure shown in Fig. 1, the orientation of Yb–O bonds and Al–O bonds in space is quite random, which may result in anisotropy in elastic behavior. To get a clear and complete representation of the elastic anisotropy of Yb3Al5O12, the variation of Young's modulus as a function of crystal orientation is investigated. The direction-dependence of Young's modulus, E , for Yb3Al5O12is given by the following equation [49]:
where s11, s12, and s44 are the elastic compliances; l1, l2, and l3 are the directional cosines of angles with the three principal directions, respectively.
The three-dimensional surface contour of Young's modulus of Yb3Al5O12in full space is shown in Fig. 2(a), and the planar projection of Young's modulus on {110} crystallographic planes, which include all three of the principal directions of a cubic crystal:<100>, <110>, and<111>, is also shown in Fig. 2(b). The elastic anisotropy of Yb3Al5O12is quite unobvious. The three-dimensional surface of Young's modulus is nearly a sphere, and the projection on {110} planes is close to a circle. The maximum and minimum of Young's modulus are 266 GPa and 246 GPa, which are along<100>and<111>direction, respectively. Young's modulus in<110>direction is 260 GPa. The minimum is only 7.5% smaller than the maximum. The isotropy of Young's modulus is also identified by Zener ratio A [50], which is defined as: A = 2 c44/( c11– c12). A = 1 is defined to indicate elastic isotropy, i.e., when c44= ( c11– c12)/2. The value of this ratio from cubic elastic constants also determines the direction for the extreme of Young's modulus (i.e., for A <1 the maximum Young's modulus would be in the<100>directions, and for A >1 the maximum would be in the<111>directions). For Yb3Al5O12, Zener ratio is 1.10, which is slightly larger than 1. This confirms the isotropy of elastic behavior of Yb3Al5O12. Interestingly, the value of Zener ratio for Y3Al5O12is 1.03, which is also slightly larger than 1. Therefore, it is reasonable to conclude that Y3Al5O12is also isotropic in elastic behavior and the maximum of Young's modulus is also along<111>directions. The isotropy in mechanical behavior of Y3Al5O12has been confirmed by previous experimental observation [9].
3Al5O12 and (b) planar projections on 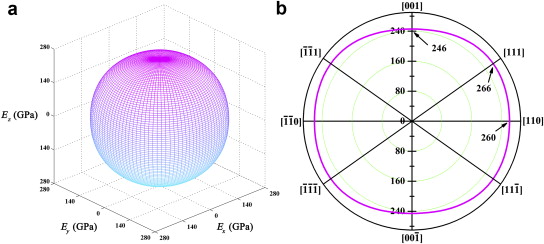 | Fig. 2. (a) Surface contour of directional dependent Young's modulus of Yb3Al5O12 and (b) planar projections on |
In the investigations and applications of a material, the orientation of grains plays a critical role in determining the mechanical performance of a material, because the anisotropy in elastic behavior generally exists, even in materials with cubic crystal structure (Zener ratio of ZrO2is 0.37–0.52, 0.44–0.59 for HfO2, 0.54–0.77 for M P2O7, 1.49 for MgO, and 2.43 for spinel) [30] and [51]. Fig. 2shows the isotropy of the elastic properties of Yb3Al5O12and also indicates that the difference of expected fracture energy in different orientations is quite limited. Thus, there is no need to consider the anisotropy of elasticity in the application of Yb3Al5O12, which is positive to promote the potential of applications as structural component and coating.
TEC is a fundamental parameter for a material. Several approaches have been proposed to estimate TEC theoretically: quasi-harmonic approximation (QHA) [52], molecular dynamics (MD) [53], CBT [40] etc. The CBT proposed by Zhang et al. [40] is a simple empirical method to predict the linear expansion coefficients of complex crystals based only on the crystal structure data. They had used this simple method to evaluate the TEC of Y3Al5O12 [40], and the estimated result showed excellent agreement with experimental data. Therefore, the linear TEC of Yb3Al5O12is evaluated by CBT in our work. The calculated results are listed in Table 6. Considering that the bond strength of Al–O bonds in AlO4 tetrahedron is substantially higher than those of the other bonds, it is not surprising that the contribution from Al–O bonds of AlO4 tetrahedron is quite smaller than those from the other bonds. The TEC of Yb3Al5O12 is 7.5 × 10-6K–1, theoretically. Experimentally, however, measurement on TEC of Yb3Al5O12has barely conducted. Compared with the theoretical TEC of Y3Al5O12, 7.98 × 10-6K–1, obtained by Zhang et al. [40], the TEC of Yb3Al5O12is slightly smaller. This can be comprehended from the structure of these two compounds. With a same coordination number of 8, the ion radii [54] of Y3+ (102 pm) is larger than that of Yb3+(98 pm) owing to the contraction of the 4f shell. With smaller ion radius, Yb–O bonds are supposed to be stronger than Y–O bonds, which would result in smaller TEC.
As shown in Eqs. (3)and (5), estimation of the intrinsic thermal conductivity of Yb3Al5O12relies on the knowledge of sound velocities ( υl, υs, υm), Debye temperature ( ΘD), and Grüneisen parameter ( γ). The sound velocities and Debye temperature are derived from obtained elastic moduli and density of equilibrium structure by Eqs. (6), (7) and (8). The calculated parameters for Yb3Al5O12are listed in Table 7, together with those of Y3Al5O12for comparison. It is reasonable that the sound velocities of Yb3Al5O12 are significantly lower than those of Y3Al5O12 since higher value of density results in smaller sound velocities. Relatively low Debye temperature of Yb3Al5O12 also suggests relatively weak atomic vibrations in the lattice due to heavy atomic weight of Yb atom, comparing with Y3Al5O12.
| Table 7. Theoretical density d (in g cm-3), sound velocities ( υl, υs, υm in km s-1), Debye temperature ΘD (in K), Grüneisen parameter γ , and minimum thermal conductivity (in W m-1 K-1) of Yb3Al5O12 and Y3Al5O12 ceramics. The Grüneisen parameter of Y3Al5O12 is calculated from the reported Poisson's ratio |
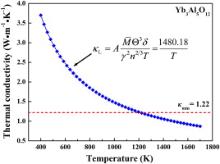
At relatively high temperature, the dominant mechanism of phonon scattering, which determines the intrinsic lattice thermal conductivity of a material, is the Umklapp processes, in which the acoustic phonon branches interact with each other to transport heat. Slack's model (i.e. Eq. (3)) [17]is a suitable approach to describe the temperature dependence of thermal conductivity when the Umklapp scattering is dominant. The temperature dependence of thermal conductivity of Yb3Al5O12estimated from Slack's model is shown in Fig. 3. The thermal conductivity of Yb3Al5O12decreases with temperature. With the temperature increases further, the thermal conductivity would approach a minimum when the phonon mean-free path decreased to the average atomic distance [15]. The minimum thermal conductivity κmin can be evaluated by modified Clarke's model, as illustrated by Eqs. (5) and (6). The minimum thermal conductivity of Yb3Al5O12 was predicted to be 1.22 W m-1K-1. Combining these two models, the behavior of thermal transportation of Yb3Al5O12 can be described as that, with increasing the temperature, the thermal conductivity of Yb3Al5O12declines as κ = 1480.18/ T , and at around 1200 K, it reaches a minimum value and then keeps unchanged with increasing temperature.
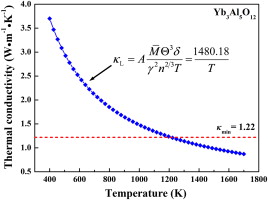 | Fig. 3. Temperature dependence of thermal conductivity of Yb3Al5O12. Theoretical minimum thermal conductivity (red dash line) is also shown. |
The minimum thermal conductivity of Yb3Al5O12is 1.22 W m-1K-1, which is 23% smaller than that of Y3Al5O12. Zhan et al. [10] theoretically investigated the thermal conductivity of Y3Al5O12. They concluded that the origin of low thermal conductivity of Y3Al5O12 was mainly attributed to the heterogeneous bond strength. The softer Y–O polyhedra are equivalent to the thermal rattle structures, and provide “weak zones” that scatter phonons and reduce phonon mean free path. These structure features are also applicable to Yb3Al5O12, Yb–O polyhedra also provide “weak zone” to scatter phonons. In addition, as suggested by Clarke [15], the strongest influence on the minimum thermal conductivity is the ratio of the atomic weight to the number of atoms per molecule, i.e. mean atomic weight M/n in Eq. (5). The mean atomic weight of Yb3Al5O12(42.3 amu/atom) is 30% larger than that of Y3Al5O12(29.7 amu/atom), which results in lower thermal conductivity of Yb3Al5O12.
The major criterion for a prospective TBC material is low intrinsic thermal conductivity. Besides, several additional selection criteria have also been proposed by Clarke [15]. First of all, TBC material must be thermodynamically stable with Al2O3 since the majority of TBCs are used in conjunction with alumina-forming alloys. For Yb3Al5O12, it is stable with alumina till they melt at around 2100 K [55]. Second, TBC material should not exhibit appreciable mass loss at high temperatures, especially in the presence of water vapor. As reported by Klemm [11], the surface corrosion rate of Yb3Al5O12 in hot gas environments is appreciably smaller than those of several other prospective TBC materials, such as Y3Al5O12, Y2Si2O7 and Yb2Si2O7. Moreover, as the weight of a coating increases linearly with both its thickness and density, a key factor affecting the design of TBC is the specific thermal conductivity, namely the product of thermal conductivity and the density. The specific thermal conductivity of Yb3Al5O12(8.14) is smaller than that of yttria-stabilized zirconia (9.31) [56], the most successful and commercialized thermal barrier material.Good stability with alumina at moisture environments and small specific thermal conductivity ensure Yb3Al5O12 a prospective candidate as coating TBC material.
In this work, bonding characteristics, elastic stiffness, and thermal conductivity of Yb3Al5O12 were investigated by first-principles calculations and CBT. The theoretical results reveal the heterogeneous bonding nature of Yb3Al5O12, i.e. Al–O bonds are stronger than Yb–O bonds. The second-order elastic constants and mechanical properties of Yb3Al5O12 were calculated. Low shear modulus G suggests shear deformation resistance of Yb3Al5O12is low and damage tolerance. The hardness of Yb3Al5O12is 14.9 GPa from calculated elastic moduli, which is consistent with the hardness (13.1 GPa) estimated from CBT. Relatively high hardness indicates poor machinability of Yb3Al5O12. Yb3Al5O12 shows isotropy in elastic behavior based on the discussion of direction dependence of Young's modulus. Using CBT, the TEC of Yb3Al5O12 is estimated to be 7.5 × 10-6K–1. The temperature dependence of thermal conductivity of Yb3Al5O12 is predicted based on the obtained elastic moduli, sound velocities and Debye temperature. The intrinsic thermal conductivity of Yb3Al5O12 decreases as 1480.18/ T and approaches a minimum thermal conductivity (1.22 W m-1K-1) at high temperatures. Weak Yb–O bonds and large mean atomic weight contribute to the low thermal conductivity. Our theoretical results highlight the potential of Yb3Al5O12as a promising TBC material.
This work was supported by the National Outstanding Young Scientist Foundation for Y.C. Zhou under Grant No. 59925208, and the National Natural Science Foundation of China under Grant No. 50832008.
| 1. |
|
| 2. |
|
| 3. |
|
| 4. |
|
| 5. |
|
| 6. |
|
| 7. |
|
| 8. |
|
| 9. |
|
| 10. |
|
| 11. |
|
| 12. |
|
| 13. |
|
| 14. |
|
| 15. |
|
| 16. |
|
| 17. |
|
| 18. |
|
| 19. |
|
| 20. |
|
| 21. |
|
| 22. |
|
| 23. |
|
| 24. |
|
| 25. |
|
| 26. |
|
| 27. |
|
| 28. |
|
| 29. |
|
| 30. |
|
| 31. |
|
| 32. |
|
| 33. |
|
| 34. |
|
| 35. |
|
| 36. |
|
| 37. |
|
| 38. |
|
| 39. |
|
| 40. |
|
| 41. |
|
| 42. |
|
| 43. |
|
| 44. |
|
| 45. |
|
| 46. |
|
| 47. |
|
| 48. |
|
| 49. |
|
| 50. |
|
| 51. |
|
| 52. |
|
| 53. |
|
| 54. |
|
| 55. |
|
| 56. |
|