Search for articles:
Yingzhi Chen, Dongjian Jiang, Zhengqi Gong, Qinglin Li, Ranran Shi, Zexi Yang, Ziyi Lei, Jingyuan Li, Lu-Ning Wang
Corresponding authors:
Received: 2019-05-7
Revised: 2019-06-12
Accepted: 2019-09-19
Online: 2020-02-01
Copyright: 2020 Editorial board of Journal of Materials Science & Technology Copyright reserved, Editorial board of Journal of Materials Science & Technology
More
Abstract
The ease of molecular design and functionalization make organic semiconductors (OSCs) unit the electronic, chemical and mechanical benefits with a material structure. The easily tunable optoelectronic properties of OSCs also make it promising building blocks and thereby provide more possibilities in photocatalytic applications. So far, organic nanostructures have gained great impetus and found wide applications in photocatalytic organic synthesis, remediation of water and air, as well as water splitting into hydrogen. But they still suffer from low charge separation and sunlight absorption efficiencies. Accordingly, many strategies have been explored to address these issues, and one of the most effective solutions is to develop nano-heterostructures. To give an impulse for the developments of this field, this review attempts to make a systematic introduction on the recent progress over the rational design and fabrication of organic nano-heterostructured photocatalysts, including the types of organic semiconductor/semiconductor (OSC/SC), organic semiconductor/metal (OSC/M), organic semiconductor/carbon (OSC/C), and OSC-based multinary nano-heterostructures. The emphasis is placed on the structure/property relationships, and their photocatalytic purposes in environmental and energy fields. At last, future challenges and perspectives for the ongoing development of OSC materials and their use in high-quality optoelectronic devices are also covered.
Keywords:
The energy shortages and environmental pollution have constitute a worldwide concern, and thus demand a ready solution to assure the sustainable development of human society. Among the various proposed solutions, semiconductor-based photocatalysis has gained increasing interest over the past few decades, because it can convert solar energy to chemical fuels, in the form of hydrogen and hydrocarbon fuels, or to address environmental issues by purification of harmful pollutants [[1], [2], [3], [4]]. Since the pioneering work on photocatalysis reported by Fujishima and Honda in 1972, many semiconductors have been explored and tested as photocatalysts [[5], [6], [7], [8], [9]]. Generally, a photocatalytic reaction mainly include five steps [10,11]: i) light absorption to form electron-hole pairs, ii) separation of electron-hole pairs and migration to the semiconductor surface, iii) adsorption of the reagents, iv) redox reaction on the semiconductor surface, v) desorption of the products. In this regard, current efforts are being made to maximize the efficiency of these processes, mainly through increasing the solar light utilization and preventing the recombination of electrons and holes. As the first requirement, visible-light response is often a must for the applicability of a photocatalyst.
Previous work has mostly concentrated on the exploration of inorganic semiconductors (ISCs) as photocatalysts, but the fixed- and narrow-spectrum response often limits its overall catalytic efficiency [[12], [13], [14]]. By comparison, the use of organic semiconductors (OSCs) constitutes an important alternative, considering their strong and wide absorption bands in the visible region [[15], [16], [17], [18]]. Another important merit concerning organic materials is that their molecular structure and photoelectric properties can be simply tuned through molecular design and tailoring. Moreover, the progresses made in the supramolecular assembly of OSCs have prompted their photocatalytic applications by enhancing the stability and possibility of reuse in heterogeneous media. The effect of shape, size, crystal type and self-assembly on the optoelectronic properties have been studied extensively for organic nanostructures. Among the candidates, porphyrins [[19], [20], [21]], phthalocyanines [[22], [23], [24], [25]], fullerenes [25,26], perylenetetracarboxylic diimide derivatives (PDIs) [27,28], g-C3N4 [29,30], and etc have stood out (Fig. 1), because they possess many exciting features like excellent semiconducting behavior, thermal stability, and the self-assembly ability into ordered nanostructures. Thus far, a series of papers have appeared dealing with nanoscale organization of them for different photocatalytic purposes, involving organic synthesis, remediation of water and air, and water splitting into hydrogen.
Fig. 1. Chemical structures of several typical organic photocatalysts: (A) Porphyrins, (B) Phthalocyanines, (C) Fullerenes, (D) PDIs and (E) g-C3N4.
Although great advances have been made in nanostructured organic photocatalysts, a single component can still not do a good job because of the short photogenerated electron-hole pair lifetime. It is widely accepted the electron-hole pairs tend to recombine very fast, i.e., in the range of nanoseconds. In order to enhance the spatial charge separation efficiency, a rational design of organic nano-heterostructures is always a research focus [[31], [32], [33]]. The heterostructures can allow for not only retarding the charge recombination, but above all going beyond the features of each one due to the synergistic effects. As a matter of fact, the design of organic nano-heterostructures provides a solid basis for creating high-efficiency photocatalysts for practical use.
Since a systematical information over the development of the field is still missing, we herein make a comprehensive review on the recent efforts in engineering various organic nano-heterostructures, with emphasis on the structure/property relationships, and their use in photocatalysis, including hydrogen evolution, CO2 reduction, and pollutant purification. Before that we give a brief introduction of their structural and optoelectronic properties. Some challenges and perspectives on the future developments are also be covered in this review.
OSCs are a class of materials that are primarily composed of carbon and hydrogen atoms, sometimes containing a few heteroatoms like nitrogen, oxygen, and sulfur. They unit the electronic, chemical and mechanical benefits with a material structure that can be easily adjusted via chemical synthesis. That is, it is possible to tune the electronic properties for desirable absorption/emission wavelength, to make it soluble/insoluble, or to render it mechanically robust, lightweight, and flexible. These properties imply that the techniques available for processing or patterning OSCs may be realized with a variety of solution-processing techniques or vacuum deposition methods [34,35]. For example, the methods for patterning organic materials can include printing, embossing, imprint lithography, and etc. These properties also enable the booming flexible applications by use of OSCs as active materials in electronic and optoelectronic devices.
OSCs have the electronic advantages typical of semiconductor materials, but differ strongly from ISCs [36]. The weak, non-covalent, van der Waals forces between organic molecules always result in a low intermolecular orbital overlap and low dielectric constant, taking a value of about εr≈3.5 [37,38]. This implies that coulomb interactions are significant enough to bind the electron-hole pairs created by optical excitation. In another word, photon absorption in OSCs affords the generation of an exciton, or coulombically bound electron-hole pair. In order to overcome the exciton binding energy for useful charge carriers, the photogenerated excitons must diffuse to a heterostructure interface that has a sufficient band energy offset. While for ISCs, free charges are created by optical excitation from a valence band (VB) to a conduction band (CB), because the high dielectric constant (εr≈11) makes the coulomb effects between electrons and holes trivial due to dielectric screening.
Compared with the electronic coupling between atoms in ISCs, the weaker coupling between molecules in OSCs also leads to relatively narrow VBs and CBs, more localized charge carriers, lower carrier mobilities (typically 10-5-0.1 cm2 V-1·s-1), along with small exciton diffusion lengths (LD ≈ 3-40 nm) [37]. As for the more localized carriers in OSCs, the terms of highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are used here to replace VB and CB, respectively. The mobility limitations also emphasize the importance of building nanoscale interfaces, where the excitons can diffuse a short distance typically on the order of tens of nanometers before relaxing.
Despite these limitations, most organic materials have high absorption coefficients of α > 105 cm-1, so at the absorption peak, a thin film with thickness of only 100 nm can absorb 90% of the light incident on it [18]. This means that the thicknesses of organic layer can be kept very thin but still highly absorptive and meantime the charge transport behavior can be preserved. As mentioned above, the absorption spectra of OSCs are broad and, in addition, can be modified by synthetic chemistry to give light absorption across the visible spectrum and even into the near infrared. A basic knowledge of these features establishes the rationale for the various device architectures, and also accounts for the rapid progress in the field of photocatalysis recently.
For the past few decades, the unique optoelectronic properties of OSCs have intrigued great exploration on large aggregation of organic molecules, which are held together mainly by noncovalent intermolecular interactions, including π-π stacking, van der Waals forces, hydrogen bonding, coordination interaction, and etc. Accordingly, various methods have been put forward to fabricate organic nanostructures with different morphologies, crystallinities, and optoelectronic properties. Reprecipitation is the most extensively employed strategy to prepare zero-dimensional (0D) organic nanostructures [39]. Self-assembly of organic molecules in liquid phase, or via vapor deposition is a key strategy for fabrication of one-dimensional (1D) structures [40]. Beyond the isolated nanostructures, the scale-up of them into higher-ordered architectures with complexity and hierarchy can also be achieved via liquid phase or vapor phase strategies, or different printing strategies. During the assembly, morphological control can be attained by a fine control over the self-assembly process. It is also noticed that the optoelectronic properties display an obvious morphology dependence in the organic aggregates during these processes.
For instance, in the case of zinc meso-tetra(4-pyridyl)porphyrin (ZnTPyP), the resultant morphologies changed from nanoparticles (NPs) to nanofibers (NFs) just by tuning the surfactant concentration or the aging time during the reprecipitation process [41]. Moreover, the 1D NFs displayed a higher catalytic efficiency in photooxidizing rhodamine B (RhB) than the 0D NPs. Similar results are also gotten from the photocatalytic water splitting test using the three photocatalysts of meso-tetra(4-pyridyl)porphyrin (H2TPyP) nanooctahedra, ZnTPyP nanowires (NWs), and an intermediate mixture of H2TPyP nanooctahedra [42]. Among them, ZnTPyP NWs exhibited the highest hydrogen evolution rate of 47.1 mmol g-1 h-1. The higher reactivity of 1D nanostructures is the result of the strengthened intermolecular π-π coupling along the long axes. The long axes of π-π stacks have extensive delocalization, hence increasing the light absorption, as well as providing a preferential guide for electron without trapping effects, which enhances the lifetime of the charge carriers.
It is highly recognized that mobility can undergo an increase up to several orders of magnitude when the molecular packing gets improved [43]. As an example, electron mobility in C60 single crystals was tested to be 2.1 cm2 V-1 s-1, but it fell by at least 3 orders of magnitude by imperfect purification, uncontrolled crystallization, or by oxygen traps [38]. Another study revealed that, impurified pentacene had a mobility of 35 cm2 V-1 s-1 at room temperature, which was promoted to 58 cm2 V-1 s-1 at 225 K in the case of pentacene single crystals [44]. With this in mind, growing effort has been devoted to the synthesis of photocatalytic nanocrystals.
For example, the Sn (IV) meso-tetraphenylporphyrin dichloride (SnTPPCl) nanocrystals were prepared via an interfacial self-assembly driven microemulsion process [45]. By adjusting the reaction conditions, different nanocrystals were obtained, such as nanosheets (NSs), octahedra, and microspheres. The hierarchical nanocrystals displayed collective optical properties due to molecular ordering, and hence excellent photocatalytic behavior in platinization and decomposition of methyl orange (MO). Further examples were the use of acid-base neutralization method to give different morphologies of ZnTPyP [46]. With increased surfactant concentration, a series of morphologies were synthesized with controllable size and dimensions, including amorphous NPs, as well as crystalline nanodisks, tetragonal nanorods (NRs), and hexagonal NRs. The crystalline and porous nanodisks showed the best performance. In the two cases, the X-ray diffraction (XRD) patterns of SnTPPCl octahedra were assigned to a tetragonal space group, whereas the ZnTPyP tetragonal NRs indexed as a monoclinic space group. Here the improved mobility was the major reason for the enhanced photoactivity.
The above concepts have been proved to be successful to boost the mobility, but still restricted to some extent. Interfacial nano-heterostructuring is a more feasible strategy to overcome the mobility limitation. The built-in energy level offset within the heterostructure provides a driving force to separate the exciton for ready charge transfer. In addition, a good control of the interfacial properties plays a critical role in achieving higher efficiency. Recently, increasing studies have been reported on the design and preparation of organic nano-heterostructures to enhance the overall photocatalytic efficiency. Typically, there are four types of nano-heterostructured organic photocatalysts, including: organic semiconductor/semiconductor (OSC/SC), organic semiconductor/metal (OSC/M), organic semiconductor/carbon (OSC/C), as well as OSC-based multinary nano-heterostructures. The latest progresses over these heterostructures are covered in detail in the following sections.
In OSC/SC heterostructures, the most applicable for photocatalysis is the staggered bandgap type (type II, Fig. 2) [11]. For type II, the CB and VB levels of semiconductor A lie above the corresponding levels of the semiconductor B. when A and B come into contact, the built-in electric field across the interface can direct the electrons and holes to flow in the opposite direction. In this case, the collected electrons in the CB of A will be transferred into the CB of B, while the left holes will migrate from the VB of B to the VB of A upon light excitation, reaching an aim to separate the spatial electron-hole pairs.
Fig. 2. Schematic depiction of the type II in the role of separation of photogenerated electron-hole pairs.
For example, we have succeeded in the design of type II organic/inorganic nanojunctions for enhanced photoelectrochemical (PEC) water splitting [47]. The junction was composed of n-type TiO2 nanotube (NT) arrays and n-type PDI organic overlayer [48]. Anodization was adopted to give TiO2 NT arrays, which had a pore diameter of about 40 nm, wall thicknesses of about 30 nm, and length up to 4 μm. The PDI overlayer was coated on TiO2 by physical vapor deposition (PVD). A control on the distance between the deposition and evaporation zones gave rise to PDI overlayer of different thickness (Fig. 3(A)), making it possible to optimize the interfaces and thus to improve its optoelectronic properties for photocatalytic use. After optimization, the charge transfer resistance could be reduced from the original 2738 Ω to 1290 Ω (Fig. 3(B)) fitted from the electrochemical impedance spectroscopy (EIS) results. The charge separation behavior was further monitored by FL emission spectra and the time-correlated single-photon counting (TCSPC) technique. A $\widetilde{9}$0% FL quench was observed for PDI after hybridizing with TiO2 and its FL lifetime was increased from 0.9 ns to a maximum 4.6 ns after hybridization. The FL measurements were in line with the EIS results, which were responsible for an improved charge separation efficiency. This improvement was well collaborated by the band alignments of PDI and TiO2 for smooth charge transfer, where TiO2 had a lower-lying CB edge (ECB =-3.8 eV) than the LUMO band of PDI (ELUMO =-4.2 eV). Furthermore, a wide visible-light response was achieved over 400-600 nm as plotted in the incident photon-to-current-conversion efficiency (IPCE) figure (Fig. 3(C)). As a consequence, the junction displayed a photocurrent value as high as 0.74 mA cm-2 at 1.23 V vs. RHE even when no co-catalyst or sacrificial reagents were added (Fig. 3(D)). This value was superior to that by most reported TiO2- or organo-based photoanodes.
Fig. 3. (A) SEM images of (a) TiO2 nanotube arrays; (b) PDI/TiO2 junction I; (c) II; (d) III; (e) IV and (f-j) their corresponding high-resolution TEM (HRTEM) images in sequence. (B) Nyquist plot at 1 V applied potential vs. RHE. (C) IPCE plots in the 400-700 nm range at 1.23 V vs. RHE. (D) Photocurrent vs. applied potential under chopped illumination. All measurements in B-D were carried out in NaOH solution (8.1 pH) under 100 mW cm-2 illumination. Adapted from Ref. [
In another report, TiO2 was coated on the PDI NFs through a facile in-situ solution deposition to get the 1D nanocomposites [27]. Of this composite, PDI worked as the major photocatalyst that responded to visible light, and TiO2 functioned as electron transfer relay to influence the overall performance by accepting electron from PDI and further transferring it to the co-catalyst Pt. Electron transfer occurred from the excited PDI to lower-lying TiO2, and then was trapped by the Pt to participate in the H2 evolution reaction. Further by side chain modification, 1D π-π stacking between PDI molecules could be adjusted to afford extensive delocalization of the electron along the long axe of the NFs. When irradiated by visible light (λ > 420 nm), hydrogen production was detected for the system by photocatalytic water splitting in the presence of sacrificial reagent methanol or triethanolamine. Besides PDI/TiO2, PDIs have been enrolled in constructing a series of heterostructured photocatalysts, for instance, PDI/ZnS [49], PDI/CdS [50], and PDI/g-C3N4 [51]. These heterostructures benefit much from PDI’s strong visible-light absorption, tunable energy levels by chemical functionalization, as well as matched energy levels. Increasing efforts have been made to tune the electron donor-acceptor structure of PDI molecules and fabricate 1D nanostructures of PDIs, so as to adjust the structural and electronic properties at the interfaces for improved charge transfer efficiency.
g-C3N4 is also an n-type OSC, and stands out as a visible-light photocatalyst due to its suitable bandgap ($\widetilde{2}$.7 eV), chemical stability, along with the ease of making thin-layer NSs and porous structures [52]. Many studies have been reported on hybridizing g-C3N4 with ISCs to construct the type II heterostructures. These ISCs covered TiO2 [53], ZnO [54], Fe2O3 [55], MoO3 [56], SnO2 [57], V2O5 [58], CdS [59], ZnS [60], MoS2 [61], Ta3N5 [62], SiC [63], BiVO4 [64], Bi2WO6 [65], Ag3PO4 [66], AgBr [67], AgI [68], and etc.
For example, we have synthesized a bimodal macroporous g-C3N4/SnO2 nanohybrid for photooxidation of methyl blue (MB) [57]. The hybrid was facilely synthesized through heat treatment of the homogeneous mixture of thiourea and SnCl4 that were dried by rotatory evaporation. The involvement of SnCl4 could not only regulate the pore structure of g-C3N4 into NTs to render it highly macroporous, but also yield SnO2 to couple with g-C3N4 for formation of charge transfer interfaces. An improvement on the macropore structures in the g-C3N4/SnO2 hybrids gave rise to an enhanced specific surface area (SSA) of 44.3 m2 g-1 (Fig. 4(A) and (B)) that could keep up with most nano/mesoporous g-C3N4 reported. As a result, the abundance of the in-plane macropores and active sites contributed to a higher light absorption, as well as a significantly increased charge separation efficiency, which was confirmed by the large quench of photoluminescence (PL) intensity (Fig. 4(C)) and higher photocurrent generation (Fig. 4(D)) for the hybrid. Mechanistic insight into the higher charge separation was gained from the band structures. SnO2 has a CB (ECB = -0.11 eV) and VB (EVB =3.59 eV) that lie below that of g-C3N4 (ECB = -1.12 eV, EVB =1.57 eV). The matched band alignment and the large SSA thus provided a great impetus to separate the photogenerated charges. Therefore, the g-C3N4/SnO2 hybrid exhibited a 2.4 times higher photocatalytic activity than pure g-C3N4 in photooxidizing MB (Fig. 4(E)), and long-term stability was also ensured for repeated use (Fig. 4(F)).
Fig. 4. (A) TEM image of g-C3N4/SnO2 Hybrid II. (B) The Brunauer-Emmett-Teller (BET) SSA of the as-prepared samples. (C) PL emission spectra. (D) The photocurrent vs. time plots at 1.23 V vs. RHE under chopped illumination. (E) Photocatalytic degradation of MB under solar light for the obtained samples. (F) Recycling experiment using Hybrid II for MB degradation under solar light. The light intensity in D-F is 100 mW cm-2. Adapted from Ref. [
As covered in most reviews, the g-C3N4 was reported to hybridize with ISCs. While, its coupling with OSCs also constitutes an important part in the photocatalytic applications, which is yet not well dealt with in the previous reviews. The structural similarity between polymeric g-C3N4 and small-molecular OSCs makes their coupling more versatile, which can either performed through π-π conjugated interaction or through covalent bonds [[69], [70], [71], [72], [73]].
In one study, g-C3N4 was coupled with a zinc phthalocyanine derivative (designate as Zn-tri-PcNc) to induce efficient photocatalytic H2 production under near-infrared light [71]. The strong intramolecular interactions came from the formation of -CO-NH- bonds between the -NH end group of g-C3N4 and the COOH groups in Zn-tri-PcNc, in addition to the π-π staking (Fig. 5(A)). Such intimate forces led to an effective charge transfer behavior, which was evidence by a series of tests like photoluminescence (PL) spectra, time-resolved photoluminescence spectra (TRPS), as well as band alignment analysis. The decreased PL emission was a good probe into the occurrence of electron transfer between g-C3N4 and Zn-tri-PcNc. In thermodynamic aspect (Fig. 5(B)), the CB level of g-C3N4 was -1.12 eV, which was sufficiency positive than the LUMO of Zn-tri-PcNc (-1.40 eV). It indicated that the electron transfer favorably took place from the excited Zn-tri-PcNc to g-C3N4. In dynamic aspect, the electron transfer process was detected by TRPS (Fig. 5(C)). The FL lifetime was calculated to be 3.97 ns for single Zn-tri-PcNc, but prolonged to be 6.23 ns after interaction with g-C3N4. The longer FL lifetime allowed the photogenerated electrons to be efficiently transferred from Zn-tri-PcNc to g-C3N4, and then trapped by the cocatalyst Pt. The cascading electron transfer finally resulted in a largely enhanced and stable visible-light photoactivity (125.2 μmol h-1) and turnover number (TON, ∼5008 h-1) for H2 production after adding proper coadsorbent (Fig. 5(D)).
Fig. 5. (A) Chemical structures of -COOH grouped Zn-tri-PcNc and g-C3N4. (B) Energy diagram of Zn-tri-PcNc/g-C3N4 system for the photocatalytic H2 evolution. (C) TRPS of the Zn-tri-PcNc/g-C3N4 system. (D) H2 evolution rate under light irradiation (λ > 500 nm).Adapted from Ref. [
In another example, tetracarboxyphthalocyanine (ZnTcPc) was covalently bonded to g-C3N4 to exhibit excellent visible-light photooxidation of phenols [72]. The efficient electron transfer enabled by their matched energy structure was responsible for the generation of three active species like ·O2-, hole, and ·OH, which were separately detected after the addition of corresponding scavengers BQ, KI and IPA. These active species could thus participate in the oxidation of phenos to give the products such as benzoic acid, hydroxybenzoic acid, and phthalic acid.
However, the type II heterostructure is not very competent to impede the ultrafast electron-hole recombination. In order to guarantee an efficient charge separation, p/n junction is often proposed. The synergy between the internal electric field and the band alignment make p/n junction more effective in prompting charge separation, transfer and prolonging the lifetime of the charge carriers. Particularly, a photocatalysis system composed of OSCs and a p/n bilayer differs apparently from conventional systems of ISCs in view of the response range in visible-light region.
Abe’s group have conducted great studies on the construction of organic p/n bilayers for PEC water splitting and pollutant removal [[74], [75], [76], [77], [78], [79], [80]]. In particular, significant efforts have been devoted to designing p/n bilayers responsive to wide or full visible-light energy, or even near-infrared energy. In one work, an organic p/n bilayer consisting of p-type zinc phthalocyanine (ZnPc) and n-type fullerene (C60) was fabricated by two-step vapor deposition, and used to reduce H+ into H2 [79]. The PEC measurement revealed an efficient evolution of H2 from ZnPc/C60 bilayer, and the action spectra for photocurrent measured at ITO/ZnPc (75 nm)/C60 (125 nm)-Pt further showed that H2 evolution took place across the full visible-light region of <750 nm (Fig. 6(A)). From the action spectral characteristics, it could be seen H2 evolution was induced by absorption of both ZnPc and C60 for ITO/ZnPc/C60-Pt electrode (Fig. 6(B)). Since a photovoltage of about 535 mV was present at the ZnPc/C60 interface, it was enough to dissociate the exciton formed within the bilayer into free electrons and holes at the p/n interface (Fig. 6(C)). Thus, the p/n bilayer was able to reduce H+ into H2 at the C60 side.
Fig. 6. (A) Action spectra of ITO/ZnPc/C60-Pt (irradiation direction: ◆, Pt-coated C60 surface; ●, back side of ITO-coated face) and ITO/H2Pc/C60-Pt (irradiation direction: ▲, back side of ITO-coated face) [
Like the case of ZnPc/C60, the full visible-light response was also found in the catalyst system composed of 29H,31H-phthalocyanine (H2Pc, p-type) and C60, or of H2Pc (p-type) and 3,4,9,10-perylenetetracarboxylic bisbenzimidazole (PTCBI, n-type). H2Pc/C60 bilayer with different thickness was studied for H2 and O2 evolution. Likewise, the action spectra for photocurrent showed that H2 evolution arose from the absorption of both H2Pc and C60, over the entire visible-light region of < 750 nm (Fig. 7(A)). While, the whiskered H2Pc/PTCBI bilayer showed an enhanced photoanodic output, because the formation of H2Pc whisker led to a rough and enlarged p/n interface (Fig. 7(B)) [81]. The H2Pc whisker was formed in a vertical direction towards the surface, to give an intensified absorption. While in the case of thick H2Pc whisker, the oxidation kinetics at the H2Pc/water interface was higher by near 3 orders of magnitude than that with planar H2Pc. The energy diagram was illustrated in Fig. 7(C) to show the electron transfer process. Upon excitation, the photogenerated electrons flew from p-type H2Pc to n-type PTCBI, and the holes moved from PTCBI to H2Pc then to the H2Pc/water interface. The sacrificial thiols then rapidly were oxidized by these holes to make electron-hole effectively separated. When H2Pc/PTCBI bilayer was deposited to flexible substrate like Nafion [82], it featured an all-organic photocatalyst, and worked efficiently for the degradation of trimethylamine, taking a value of ca. 40% of external quantum efficiency for CO2 formation.
Fig. 7. (A) Action spectra for photocurrent at ITO/PTCBI/H2Pc-A(●), ITO/PTCBI/H2Pc-B (▲), and ITO/PTCBI/H2Pc-C (◼). Concentration of thiol = 5 × 10-3 mol·dm-3 (pH = 10); applied potential = 0 V. (B) TEM images of ITO/PTCBI/H2Pc-B. (C) Energy diagram using the ITO/PTCBI/H2Pc system as the photoanode for water splitting. Adapted from Ref. [
In another example, the organic p/n bilayer composed of PTCBI and p-type lead phthalocyanine (PbPc) was demonstrated to be responsive to widespread photoenergy in the visible-near infrared regions of 400-1100 nm. Such wide response came from the absorption of both PTCBI and PbPc [83]. In addition, a photovoltage of ca. 400 mV accounted for the occurrence of charge separation at the PTCBI/PbPc interface, considering that the potentials of PTCBI and PbPc were 0.05 V and +0.34 V, respectively. The wide photoresponse and the photovoltage thus induced a series of photophysical reactions where FeII(CN)64- was oxidized and H+ was reduced into H2.
In our work, PVD method was adopted to give 1D organic single-crystal p/n nano-heterostructure, comprising of p-type H2TPP and n-type N,N-(dicyclohexyl) perylene-3,4,9,10-tetracarboxylic diimide (CH-PTCDI) [84]. The advantages of this p/n nano-heterostructure could be supported by the following aspects: a more effective charge separation driven by the large donor-acceptor interface; directed charge transport along the 1D long-range axes; increased mobility by crystallization; and wide and strong absorption by both H2TPP and CH-PTCDI. SEM (Fig. 8(A)) and TEM images (Fig. 8(B)) revealed the double-layer morphology where H2TPP layer had a thickness of ca. 500 nm, and the thickness of CH-PTCDI layer was ca. 200 nm. The marked fluorescence (FL) quench of CH-PTCDI well proved the efficient electron transfer between CH-PTCDI and H2TPP when they were combined (Fig. 8(C)). This was thermodynamically supported by their matched band structures, inducing the electron to flow from the excited H2TPP to CH-PTCDI, and the holes vice verse (Fig. 8(D)). The properties were responsible for the final enhancement on the photoactivity in degrading MB (Fig. 8(E)). The stability was also ensured for successive uses (Fig. 8(F)).
Fig. 8. (A and B) SEM and TEM images of H2TPP/CH‐PTCDI nanoheterojunctions. (C) FL spectra of H2TPP (P), CH‐PTCDI (N), and H2TPP/CH‐PTCDI (p/n) nanostructures. (D) Schematic illustration of the energy structure of H2TPP/CH‐PTCDI p/n system for photoexcited charge transfer. (E) Photocatalytic degradation of MB with different samples under visible light irradiation (λ > 400 nm). (F) The repeated runs for photodegrading MB using the p/n system. Adapted from Ref. [
An alternative method has also been proposed by designing OSC/M heterostructures to construct heterogeneous interfaces. When they come into contact, the potential difference will be created due to their different work functions, which drives the exciton to dissociate into free charge carriers. The potential difference here is called Schottky barrier. Moreover, the metallic NPs are also appealing for their unique catalytic, optical and electric characteristics, which can coordinate with OSCs to improve overall photocatalytic performance. The catalytic properties of some noble metals (e.g., Pt, Ru) make them an efficient cocatalyst. The surface plasmon resonance (SPR) effect is an important optical characteristic of some metallic NPs (e.g., Au, Ag), which contribute to a large enhancement of light absorption.
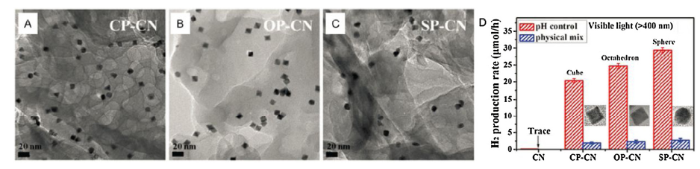
The low overpotential for water reduction makes Pt the most efficient cocatalyst for OSCs, and the Schottky barrier between them can promote the electron transfer from OSCs to Pt. Many reports have appeared dealing with Pt as cocatalyst to help enhance the photocatalytic H2 evolution. Yu’s group made great use of Pt NPs as cocatalysts to enhance the photoactivity of g-C3N4 in CO2 reduction and H2 evolution [85,86]. He also pointed out that the shape of Pt NPs had an important impact on the catalytic activity and selectivity. As in one work, different shapes of Pt NPs were precisely synthesized to have different exposed facets, and were then ex situ anchored onto the surface of g-C3N4 (Fig. 9(A)-(C)) [86]. The photocatalytic H2 evolution experiments were conducted to test its visible-light response, in which 10 vol% TEOA was added as the sacrificial agent. The photoactivity of the obtained g-C3N4-Pt hybrids was compared, following an increasing order of g-C3N4-cubic Pt<g-C3N4-octahedral Pt<g-C3N4-spherical Pt (Fig. 9(D)). As the light absorption and FL emission didn’t show much difference by changing the shape of Pt cocatalysts, the reason for the catalytic difference could be explained by different crystallographic facets. It is thought that defects are more likely to occur at some places like edges, steps or corners, and the atoms at these defect sites are more thermodynamically active to initiate physicochemical reactions. In another word, the catalytic reactivity is largely determined by the atoms at these defect sites. For instance, the (100) surface has a square atomic arrangement, which differs from (111) surface that has a hexagonal atomic arrangement. Such difference greatly affects the number of surface atoms at the defect sites, which is thereby responsible for the catalytic difference.
Fig. 9. TEM images of Pt nanocubes (CP, A), Pt nanooctahedra (OP, B) and Pt nanospheres (SP, C) supported on layered g-C3N4. (D) Photocatalytic hydrogen evolution activities of bare g-C3N4, as-prepared Pt/g-C3N4 photocatalysts and physically mixed samples under visible light within 1 h. Adapted from Ref. [
Another interesting example can be seen with porphyrin nanostructures, as their self-metallization ability provides a facile means of preparing porphyrin-metal heterostructures. This is simply done by exposing the mixed solutions of porphyrin nanostructures, a proper metal complex along with a sacrificial electron donor to visible light irradiation. On this basis, Shelnutt’s group has succeeded in obtaining a series of self-platinized porphyrin nanostructures, including NPs [87], NTs [88], NSs [89] and NFs [90], four-leaf clover-like [91]. As a matter of fact, these self-platinized porphyrin nanostructures were demonstrated to be promising visible-light photocatalysts for hydrogen evolution.
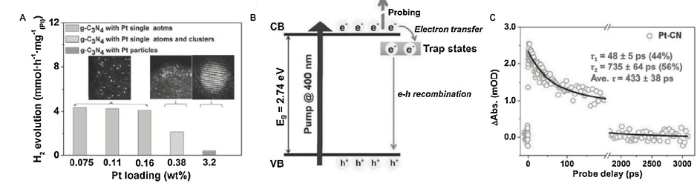
When the noble metals are downsized to clusters or even to single atoms, we can get a maximum utilization of these atoms for an enhanced photocatalytic reactivity. But the uniform dispersion of single-atom catalysts is often a difficult task because these atoms suffer from serious agglomeration with a loss of efficiency during repeated reactions. In this regard, Li et al reported the use of isolated single Pt atoms as co-catalysts to couple with g-C3N4 (Pt-CN) [92]. The reaction between the tri-s-triazine units in g-C3N4 and H2PtCl6 resulted in a rich formation of Pt-N/C bonds, making it possible to synthesize single-atom Pt-CN system under relatively low temperature. The single Pt atoms had a uniform distribution and configuration on g-C3N4. g-C3N4 with 0.38 wt% of Pt clusters exhibited a moderate photoactivity in H2 generation, g-C3N4 with Pt nanoparticles (Pt-NPs-CN) gave the worst activity. By comparison, the turnover frequency of Pt-CN (0.16 wt% Pt loading) reached 775 h-1, approximately 9 times higher than that of g-C3N4 with Pt NPs (Fig. 10(A)). The remarkable enhancement could be ascribed to the presence of the isolated single Pt atoms that caused a change on the surface trap states of g-C3N4 (Fig. 10(B)). This was elucidated by the ultrafast transient absorption (TA) spectroscopy (Fig. 10(C)), a technique to track the the real-time photoexcited carrier dynamics. The average recovery lifetime for Pt-CN was determined to be ca. 433 ps, which was twice longer than the ca. 117 ps for Pt-NPs-CN. The lengthened lifetime of the charge carriers hence revealed the modulated electronic structure of g-C3N4 due to the involvement of the single Pt atoms. Yu’s group also gained a deep insight into the mechanisms of single-atom Pt in the performance enhancement through studying the single-atom Pt-embedded g-C3N4 system.
Fig. 10. (A) Photocatalytic activity displayed by g-C3N4 with various Pt contents. (B) Schematic illustration of the involved mechanisms. (C) Ultrafast TA kinetics to probe the charge separation at 750 nm (pump at 400 nm) for Pt-CN. Adapted from Ref. [
Besides the function as an electron relay, the novel metallic NPs could absorb light to generate a strong electric field and a high concentration of hot electrons the NP surface (called SPR effect). The electrons harvest light energy and then flow to the CB of the semiconductors to fulfill the electron transfer. Thus, these plasmonic metallic NPs have been widely anchored onto the ISC or OSC support to enhance the sunlight utilization and charge separation efficiency. Plamonic Au NPs are especially used to construct plasmonic photocatalysts due to its strong visible-light absorption. For example, a calcination-photodeposition technique was used to construct plasmonic photocatalysts of Au/Pt/g-C3N4 for antibiotic degradation [93]. TEM images revealed that the spherical Au and Pt NPs with a size of 7-15 nm were uniformly formed on the surfaces of g-C3N4 (Fig. 11(A)). It was seen from the UV-vis diffuse reflectance spectra that, g-C3N4 alone displayed an absorption edge shorter than 460 nm, whereas a broadened absorption peak was observed for both the Au/g-C3N4 and Au/Pt/g-C3N4 samples at around 550 nm arising from SPR effect of Au NPs (Fig. 11(B)). When tested for photodegrading tetracycline hydrochloride (TC-HCl), a representative antibiotic, the catalytic rate followed an order of Au/Pt/g-C3N4 (93.0%) > Au/g-C3N4 (78.6%) > Pt/g-C3N4 (67.2%) > g-C3N4 (52.8%) after the same irradiation time (Fig. 11(C)). Here, the performance enhancement could be attributed to the electronic and catalytic benefits from the metallic NPs, where the SPR effect of Au resulted in an extended responsive photoenergy range, and the electron-sink function of Pt gave rise to a promoted charge separation efficiency (Fig. 11(D)). Apart from Au, Ag and some other non-nobel metals like Bi are also capable of displaying SPR effect in the visible-light region, which have been applied in SPR-enhanced photocatalysis.
Fig. 11. (A) TEM images of Au/Pt/g-C3N4 nanocomposites. (B) UV-vis diffuse reflectance spectra, and (C) Photocatalytic activities in degrading TC-HCl under visible light irradiation for the as-prepared g-C3N4, Pt/g-C3N4, Au/g-C3N4, and Au/Pt/g-C3N4 nanocomposites. (D) Schematic illustration of the catalytic mechanism for photodegrading TC-HCl by Au/Pt/g-C3N4 nanocomposites under visible light irradiation. Adapted from Ref. [
Hybridization of semiconductors with carbon materials is another way to enhance the photoenergy conversion. Among the candidates, carbon nanofibers (CNFs), carbon nanotubes (CNTs), and graphene are the most widely employed supports owing to their pleasant physical and electrical properties. The advantages of the carbon materials lie in the following aspects: i) the good conductivity renders them good acceptors and transfer/transport pathways, which leads to increased charge separation and transport efficiency; ii) the high SSA enable plentiful of active sites for the adsorption of the reactive species; iii) the similar π-conjugated and flexible structure between these carbon supports and OSCs afford them more compatible and diverse in coupling.
Graphene sheets have been highly recognized as an ideal two-dimensional (2D) scaffold due to its extremely high theoretical SSA ($\widetilde{}2$600 m2 g-1), promising charge transfer properties and tunable band-gap energy [94]. In some cases, porphyrin nanoassemblies have been included onto graphene scaffold for photocatalytic applications. In one case, Rajesh et al have fabricated a composite made of 5, 10, 15, and 20-tetrakis (4-carboxyphenyl) porphyrin (TCPP) NRs and reduced graphene oxides (rGO) to facilitated the spatial charge separation [95]. The strong coupling between rGO and TCPP NRs came from the electrostatic interaction between the positively charged NRs and negatively charged surface of the rGO. An electron transfer process was found for the composites and the electron transfer rate was estimated to be 10.0 × 10-4 ps-1. As a result, 1.9 fold enhancement of photocurrent was obtained for the composites under visible light illumination compared with that of pure TCPP NRs. Duong et al also reported the facile preparation of TCPP NRs well-dispersed on the surface of graphene nanoplates [96]. The resulted composites were also demonstrated to be good visible-light photocatalysts in degrading RhB. Another report by Guo et al has studied the inclusion of 1D porphyrin nanoassemblies on graphene oxide (GO) as photocatalysts, and tested its visible-light response [97].
We have integrated meso-tetra(p-hydroxyphenyl) porphyrin (p-THPP) NPs into rGO to get a monolithic p-THPP/rGO nanohybrid film (Fig. 12(A)) [98]. Because of coupling, the average FL lifetime of p-THPP was prolonged from ca. 362 to 473 ps (Fig. 12(B)), and meantime the charge transfer resistance was decreased from 176.2 to 46.7 Ω (Fig. 12(C)). These results indicated a greatly increased electron transfer in the hybrids, which was the main reason for the performance improvements (Fig. 12(D)). It was noteworthy that, the free-standing feature made the repeated use of p-THPP/rGO film much easier, and only a slight decrease was observed in the recycling experiment after seven repeated runs.
Fig. 12. (A) TEM image of p-THPP/rGO nanohybrids. (B) The FL decay profiles of p-THPP and p-THPP/rGO nanohybrids in H2O (λexcitation = 405 nm). (C) Nyquist plots collected by electrical impedance spectroscopy (EIS) of free-standing rGO and p-THPP/rGO films. (D) Photocatalytic degradation of MB using different samples under visible-light irradiation (λ > 400 nm). Adapted from Ref. [
The similar 2D network and π-conjugated structure between graphene and g-C3N4 have inspired the great work on their coupling into 2D/2D hybrid heterojunction [99]. Liao et al have employed sonochemical approach to modify g-C3N4 with GO overlayer, to give efficient photocatalytic capability under visible light irradiation. Ong et al have made use of rGO and protonated g-C3N4 (pCN) to construct 2D/2D heterojunction photocatalyst with effective interfacial contact [100]. The synthesis was done by combing ultrasonic dispersion, electrostatic self-assembly with a subsequent NaBH4-reduction process. Another example was provided by the formation of the 3D porous g-C3N4/GO aerogel (CNGA). The aerogel was synthesized via hydrothermal coassembly of g-C3N4 and GO NTs. The highly interconnected porous network of CNGA afforded plenty of pathways for rapid mass transport, large reactant adsorption as well as multireflection of incident light (Fig. 13(A)). In addition, the large interfacial contact between g-C3N4 and GO NTs enabled a highly promoted electron transfer (Fig. 13(B) and (C)). The resulted CNGA aerogel thus exhibited excellent photocatalytic activities for MO removal, as well as for the CO2 reduction (Fig. 13(D)) under visible-light irradiation.
Fig. 13. (A) TEM image of 15rGO/CN nanocomposites. (B) PL spectra of pCN and a series of rGO/pCN photocatalysts with different rGO contents. (C) Mechanistic illustration of the charge behaviors in the rGO/pCN nanocomposite for CO2 reduction under visible light irradiation. (D) Total evolution of CH4 over the obtained samples under visible light irradiation for 10 h. Adapted from Ref. [
1D CNFs are also a good candidate as electron transfer/transport channels. Electrospun CNFs are appealing for their monolithic feature, good conductivity (ρ=(3-7) × 10-3 Ω cm), as well as the high SSA due to its richly porous structure. Many studies have revealed that CNFs are able to capture and transport the photogenerated electrons along the 1D CNFs [[101], [102], [103], [104]]. In one study, CNF/g-C3N4 composite photocatalysts were synthesized via a two-step approach including electrospinning and a followed calcination process [105]. HRTEM image (Fig. 14(A)) indicated the existence of g-C3N4 along the surface of CNFs. The interfacial interaction between CNF and g-C3N4 was further evidenced by the strong PL quench (Fig. 14(B)). The involvement of CNFs rendered the composites a SSA of 35.9 m2 g-1 and pore volume of 0.17 cm3 g-1, which were much higher than those of pure g-C3N4 (11.9 m2 g-1 and 0.1 cm3 g-1). Because of the synergic effects of increased SSA, pore volume and charge separation efficiency (Fig. 14(C)), the CNF/g-C3N4 composites had a photocatalytic hydrogen production rate as higher as 1080 mol h-1 g-1 under visible light irradiation (≥420 nm), which was about 4.6 times higher than that of pure g-C3N4 (Fig. 14(D)).
Fig. 14. (A) HRTEM images of CF/C3N4 composite. (B) PL spectra of CF0 and CF10 (λexcitation = 360 nm). (C) Transient photocurrent responses of CF0 and CF10 electrodes in 0.5 M Na2SO4 aqueous solution under visible light irradiation. (D) Comparison of the photocatalytic H2 evolution rate. The product by addition of 0.005 g, 0.01 g, 0.015 g, 0.02 g and 0.025 g CF into 5 g of thiourea was labeled as CF5, CF10, CF15, CF20 and CF25, respectively. Adapted from Ref. [
Amorphous carbon and carbon spheres can also work as good cocatalysts to improve the photocatalytic activity. Examples can be seen with the amorphous carbon/g-C3N4 nanocomposites, and g-C3N4-encapsulating carbon spheres [106]. Likewise, the introduction of them can contribute greatly to higher visible-light absorption and larger SSA, hence benefiting the photocatalytic performance.
Core to photocatalysis is the multistep electron transfer in the photocatalysts. A well-organized multistep electron transfer system can allow for efficient photoenergy conversion. To this end, various attempts have been made in constructing ternary or multinary heterostructures on the basis of visible-light absorption, energy band match, as well as the more decisive electron transfer cascade [65,103,107].
For example, Hayashi et al have developed a multistep electron transfer system made of ternary composites (porphyrin, ZnO NPs, and rGO) [108]. The photocurrent density by this hierarchical electron transfer system was remarkably enhanced with an IPCE value as high as 70%. The difference between the LUMO of TCPP to the CB of ZnO provided an impetus for charge separation, and moreover rGO worked as an electron mediator that could accept electrons from ZnO or porphyrin moiety, hence resulting in higher photocurrent generation. Following the same strategy, Duong et al have reported the fabrication of a ternary nanohybrid using graphene/TiO2/TCPP, and it was tested to be an efficient and stable catalyst for photooxidation of RhB under solar light [109]. The SEM (Fig. 15(A)) and TEM (Fig. 15(B)) images showed that TCPP NRs and TiO2 NPs were successfully formed on the graphene surfaces. In this system, TCPP NRs absorbed visible light, and TiO2 NPs could make use of UV light to generate electron-hole pairs (Fig. 15(C)). The wide light utilization and the multistep electron transfer processes finally made this ternary system highly efficient and durable in dye photodegradation (Fig. 15(D)). Further examples are the CdTeSe-porphyrin-rGO hybrid system, to give 240-fold increase on photocurrent intensity under visible light irradiation. In this case, electrons were generated by the major photoabsorber porphyrin, and then were transferred from the LUMO of porphyrin to the CB of adjacent CdTeSe NRs due to the potential difference, and finally flowed to RGO for a cascading electron transfer.
Fig. 15. (A) SEM and (B) TEM images, (C) Proposed mechanism of photocatalytic activity, and (D) The recycle experiments for RhB degradation by graphene/TiO2/TCPP hybrid. Adapted from Ref. [
We have extended the cascading electron transfer system to the free-standing organic/inorganic ternary nanohybrids (CNF, ZnO NRs, porphyrin) [103,104,110]. ZnO has a low-lying CB edge (ECB = -4.2 eV) relative to the LUMO band of TCPP (ELUMO = -3.6 eV), that all lie above the Fermi level of CNFs ($\widetilde{-}$4.50 eV). Cascading band positions are thereby established across them when coupling. Under light illumination, excitons are generated in TCPP, and the potential gradient across the three components provides the driving force to separate the excitons and transfer the electrons from TCPP to ZnO and then to CNFs in a cascading way. The electron transfer cascade system was responsible for an enhanced photocurrent generation, and also worked more efficiently in photodegrading RhB.
Spatial integration of two or more light absorber and an electron relay into a multinary heterostructure is also a strategy to enhance solar energy conversion, since a collective light absorption from them can give rise to a broadened responsive light range. Li et al have demonstrated a facile synthesis of organic/inorganic ternary nanohybrids made of tin(IV) prophyrin (SnPor), Ag NPs, and rGO (Fig. 16(A)) [111]. The UV/Vis absorption spectra of the ternary hybrid combined the Soret band of SnPor, the absorption band of rGO, and the plasmon resonance band of Ag NPs (Fig. 16(B)). The nanohybrids exhibited high photocurrent generation (Fig. 16(C)) and good catalytic activity in the degradation of RhB (Fig. 16(D)), as a result of the photosensitization effect of SnPor and the SPR excitation in the AgNPs. A similar methodology has been employed to fabricate ternary nanocomposites of Sn-porphyrin (SnP), angular Au NPs and rGO [112]. In this system, SnP and Au NPs served as photoactive, visible-light absorbers, and the synergistic effect of SPR excitation in the AuNPs and SnP led to a 27-fold enhancement in the photocurrent density when irradiated at 550 nm.
Fig. 16. (A) TEM image of the SnPor/Ag NPs/rGO nanohybrids (inset: HRTEM image). (B) UV-vis absorption spectra of the samples: A-E in it represented GO, SnPor, SnPor/GO, SnPor/rGO, and SnPor/Ag NPs/rGO, respectively. (C) Transient photocurrent shown by different ohybrids (λexcitation>400 nm, 0 V vs. Ag/AgCl). (D) Kinetics of photodegradation of RhB (▼) by using AgNPs (▲), SnPor/rGO (●), AgNPs/RGO (★), and SnPor/Ag NPs/rGO (◼) as a photocatalyst under visible light (λ > 400 nm). Adapted from Ref. [
To sum up, this paper has outlined the recent advances on the organic nano-heterostructured photocatalysts. OSCs unit the electronic, chemical and mechanical benefits with a material structure that are easily tuned via molecular design. Assembly of them into nanostructures can lead to an improved photostability and light utilization. But the intrinsic low charge mobility still limits the overall photoconversion efficiency. Interfacial heterostructuring seems a logical step forward by prompting the charge transfer/transport process, in particular when the ease of molecular functionalization is concerned, making it convenient to adjust the interfacial or surfacial properties. This paper discussed the general strategies in making organic nano-heterostructures for developing highly efficient and stable photocatalysts. They are mainly divided into four types of OSC/SC, OSC/M, OSC/C, and OSC-based multinary nano-heterostructures, in different mechanisms to enhance the light utilization, charge transfer, and mass transport.
Despite these efforts, there are several challenges to be tackled in future. First, compared with the inorganic nano-heterostructured photocatalysts, the generation of free charges is still limited, as the size of organic nanostructures is usually larger than the inorganic counterparts so that the generated charges tend to recombine before they reach the interface. Meanwhile, the organic nanostructures are organized mostly via π-π stacking, and the weak interactions are not favorable for perfect molecular packing. So, it is commonly hard to get a good crystallization of organic nanostructures, which is not advantageous for the electron transport either. Second, the interface geometry and properties need to be clarified, and an in-depth insight into them will help to understand and determine the roles of the different components. Moreover, a quantitative prediction of the charge behaviors requires more rational calculations and simulations, especially in light of the electronic properties, excitation dynamics, and charge transport of the heterostructures. Even more importantly, the question whether the heterostructured materials are useful in the devices requires more exploration of the spatial geometry. After these issues are well addressed, the organic nano-heterostructured photocatalysts will show more promise in photocatalytic pollutant removal or water splitting by use of a wide or even full spectrum of sunlight, and they will show more feasibility in the device fabrication and environment adaptability, either in portable or deformable shapes.
The authors declare that they have no conflict of interest.
This research was financially supported by the National Key Research and Development Program of China (2016YFB0700300), the National Natural Science Foundation of China (Grant No. 51503014 and No. 51501008) and financial support from Fundamental Research Funds for the Central Universities (No. 230201818-002A3)

 WeChat
WeChat
/
| 〈 |
|
〉 |














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}